
Mapping and Detection of Genes Related to Trichome Development in Black Gram (Vigna mungo (L.) Hepper)



Abstract
1. Introduction
2. Materials and Methods
2.1. Mapping Population Construction and Sampling
2.2. Phenotype Analysis
2.3. Genomic Resequencing by Illumina
2.4. Single-Nucleotide Polymorphism Calling and Bin Map Construction
2.5. QTL Mapping and Candidate Gene Identification
2.6. Validation of Candidate Genes by Real-Time Quantitative PCR
3. Results
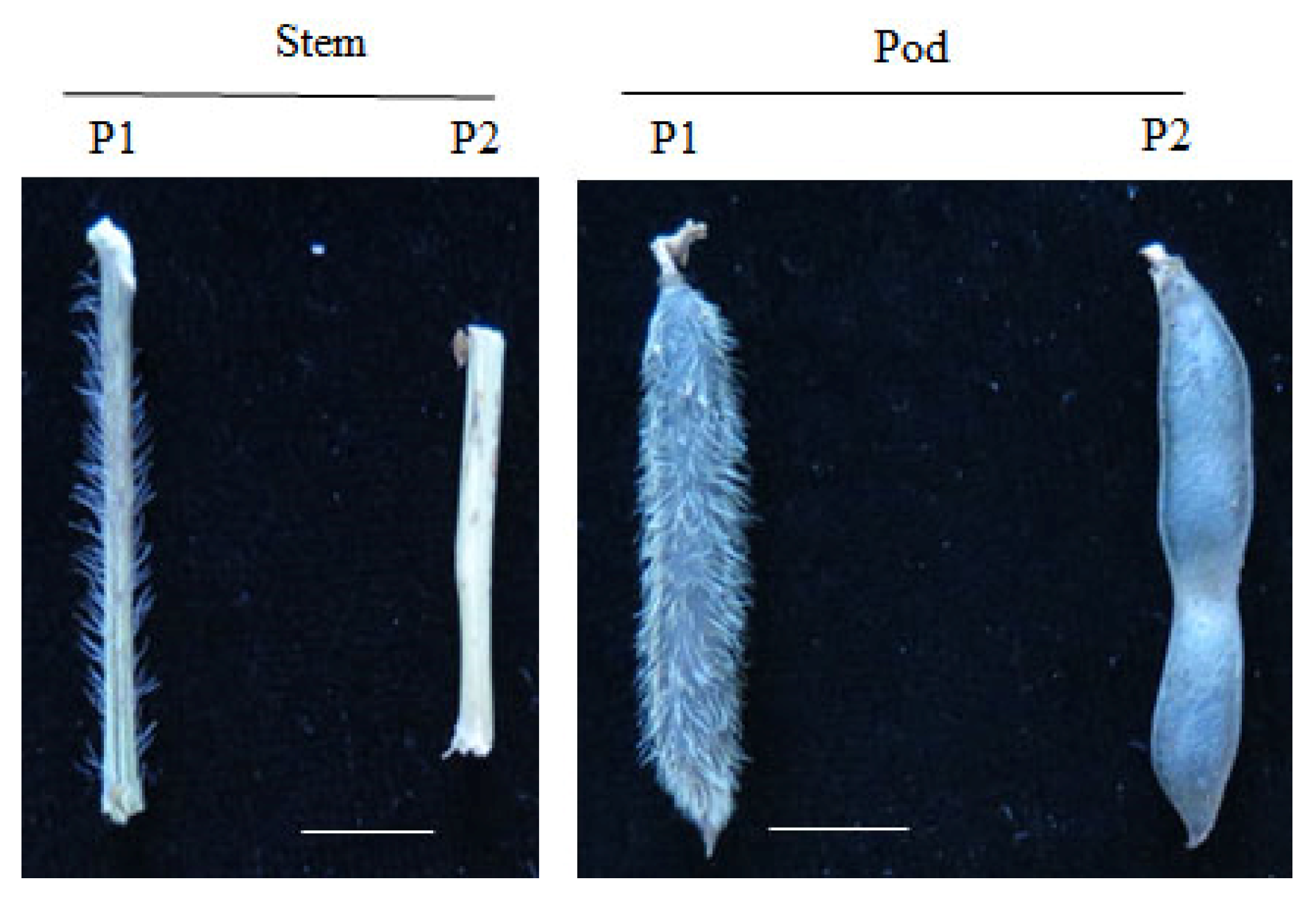
3.1. Phenotypic Variations among the Parents and the F2 Population
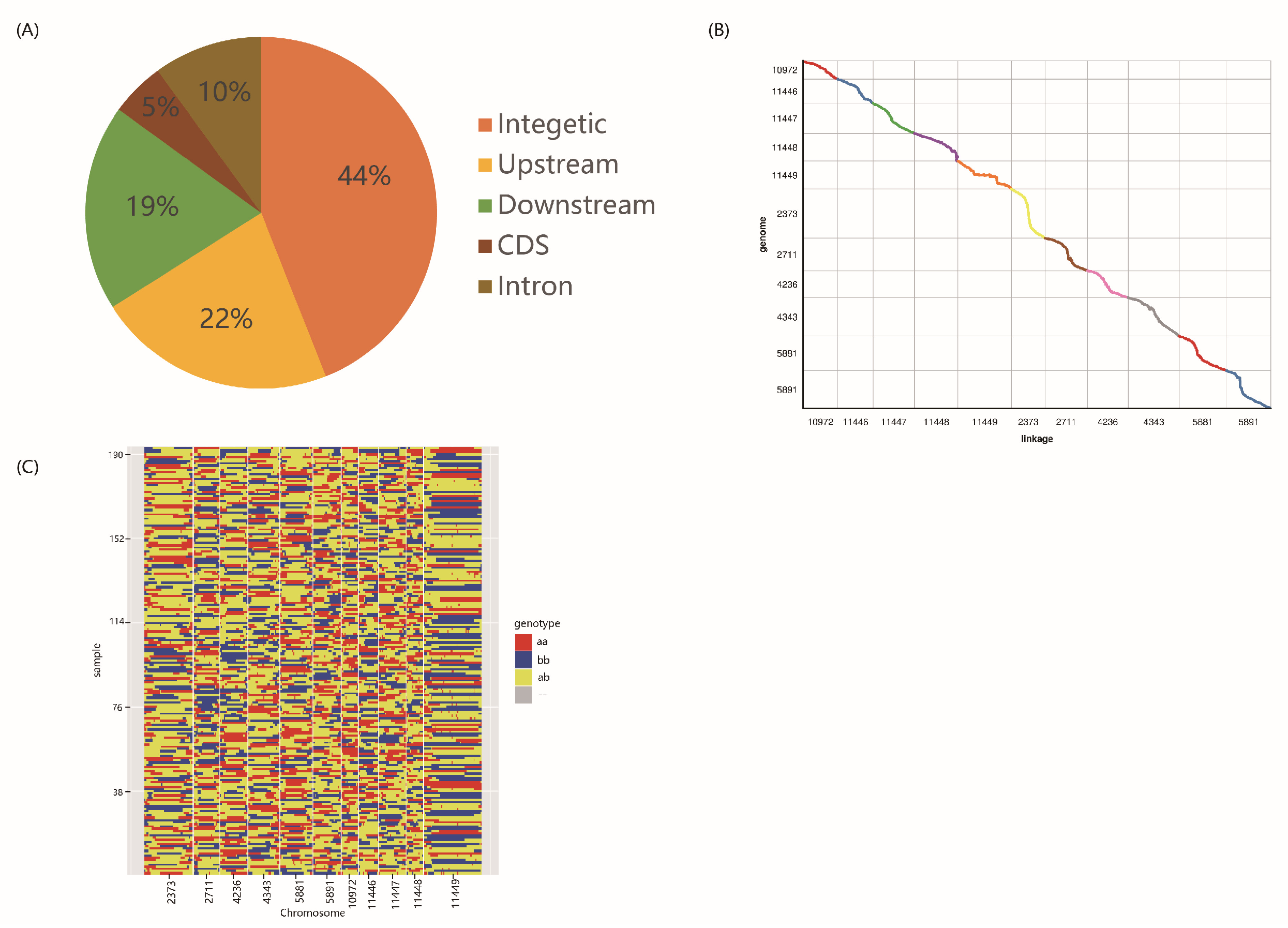
3.2. Sequencing, SNP Identification, and Bin Map Construction
3.3. Construction of Physical Recombination Maps and High-Density Genetic Linkage Map
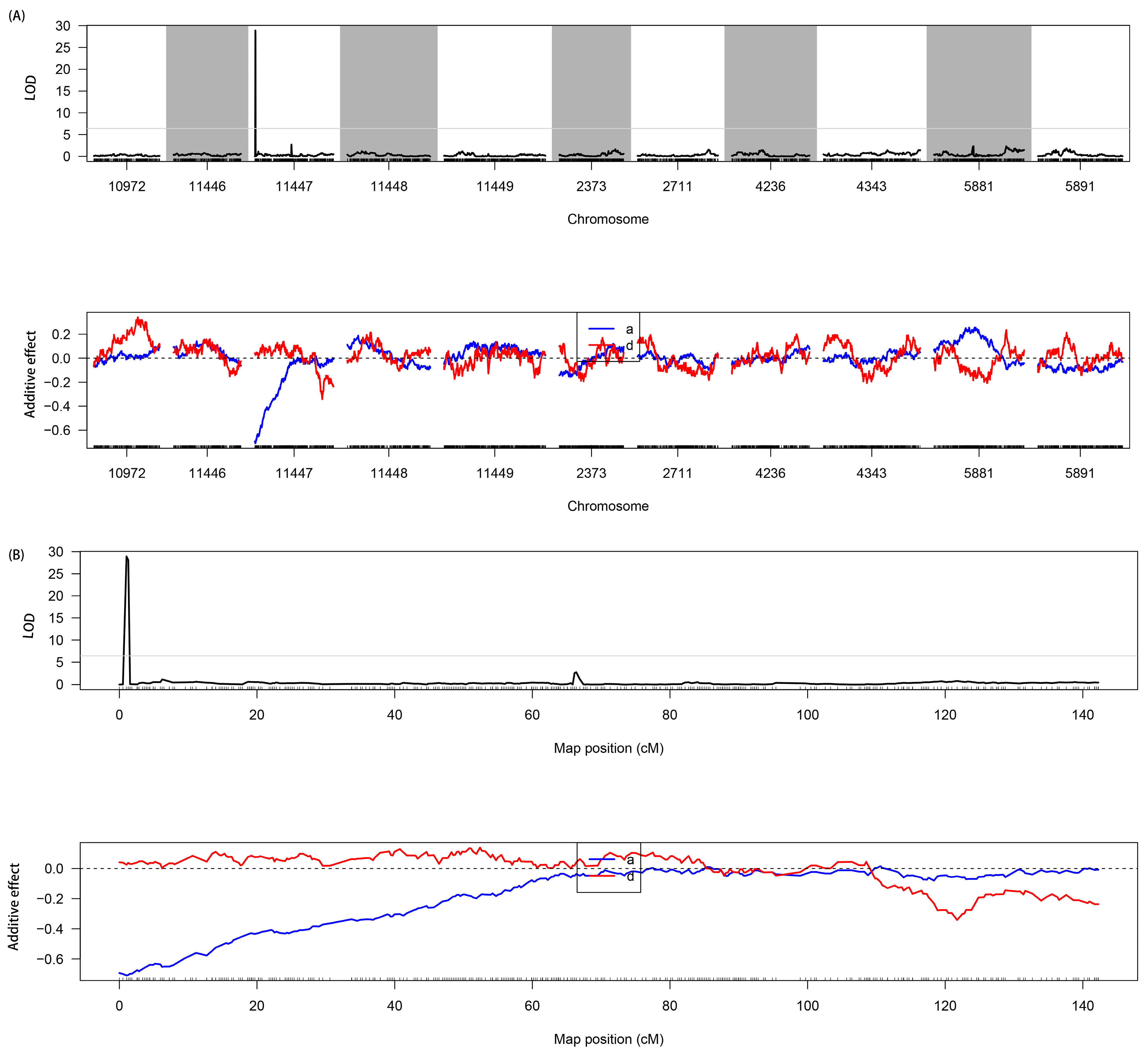
3.4. QTL Mapping and Prediction of Candidate Genes
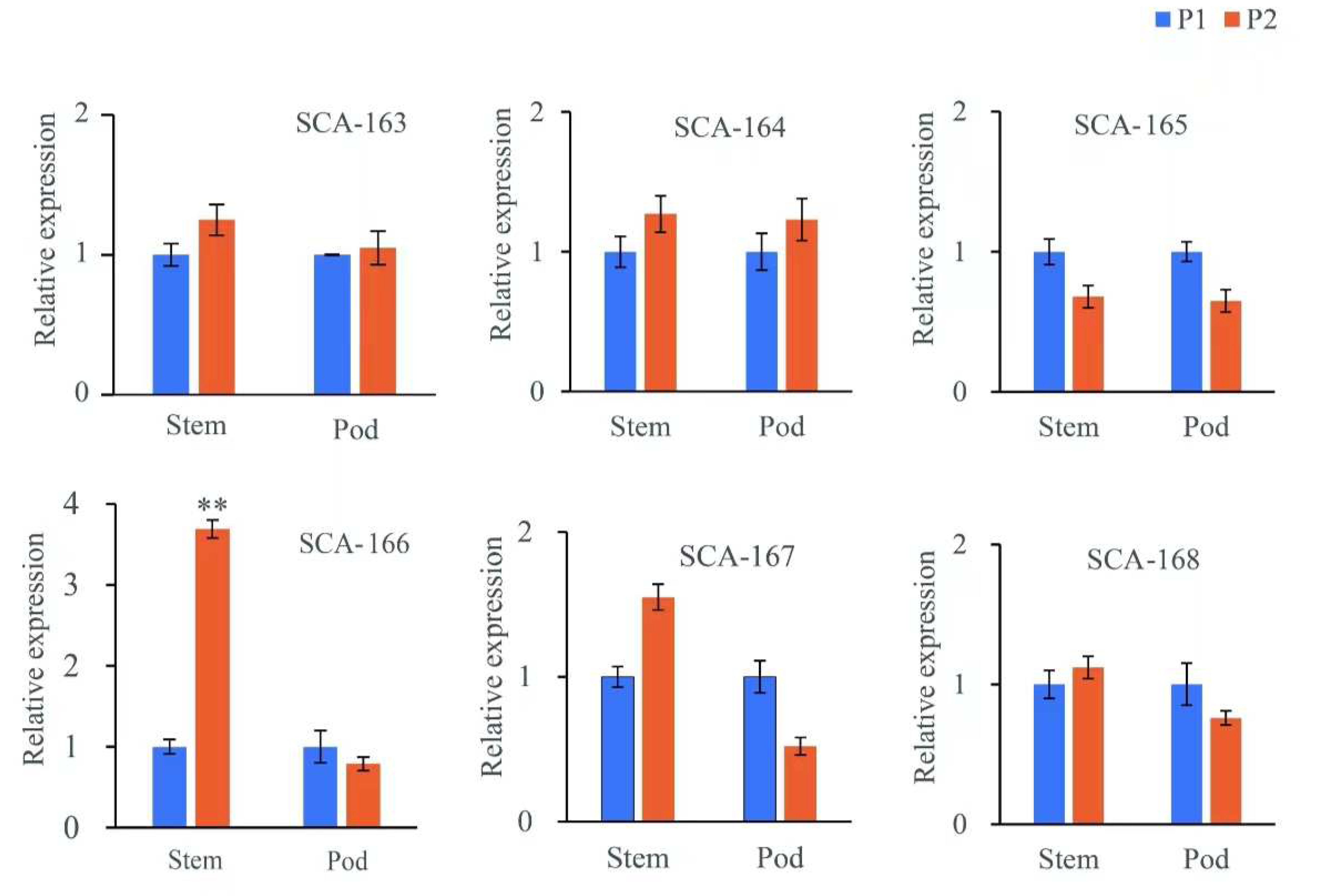
3.5. qPCR Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, H.; Zhou, L.H.; Jiao, J.; Liu, S.; Zhang, Z.; Lu, T.J.; Xu, F. Gradient mechanical properties facilitate arabidopsis trichome as mechanosensor. ACS Appl. Mater. Interfaces 2016, 8, 9755–9761. [Google Scholar] [CrossRef]
- Lloyd, A.M.; Schena, M.; Walbot, V.; Davis, R.W. Epidermal cell fate determination in Arabidopsis: Patterns defined by a steroid-inducible regulator. Science 1994, 266, 436–439. [Google Scholar] [CrossRef]
- Chien, J.C.; Sussex, I.M. Differential regulation of trichome formation on the adaxial and abaxial leaf surfaces by gibberellins and photoperiod in Arabidopsis thaliana (L.) Heynh. Plant Physiol. 1996, 11, 1321–1328. [Google Scholar] [CrossRef]
- Zeng, J.; Yang, L.; Tian, M.; Xie, X.; Liu, C.; Ruan, Y. SDG26 Is Involved in Trichome Control in Arabidopsis thaliana: Affecting phytohormones and adjusting accumulation of H3K27me3 on genes related to trichome growth and development. Plants 2023, 12, 1651. [Google Scholar] [CrossRef] [PubMed]
- Camoirano, A.; Arce, A.L.; Ariel, F.D.; Alem, A.L.; Gonzalez, D.H.; Viola, I.L. Class I TCP transcription factors regulate trichome branching and cuticle development in Arabidopsis. J. Exp. Bot. 2020, 71, 5438–5453. [Google Scholar] [CrossRef] [PubMed]
- Schuurink, R.; Tissier, A. Glandular trichomes: Micro-organs with model status? New Phytol. 2020, 225, 2251–2266. [Google Scholar] [CrossRef] [PubMed]
- Barba, P.; Loughner, R.; Wentworth, K.; Nyrop, J.P.; Loeb, G.M.; Reisch, B.I. A QTL associated with leaf trichome traits has a major influence on the abundance of the predatory mite Typhlodromus pyri in a hybrid grapevine population. Hortic. Res. 2019, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Baron, H.; Alzate, J.F.; Ambrose, B.A.; Pelaz, S.; González, F.; Pabón-Mora, N. Comparative morphoanatomy and transcriptomic analyses reveal key factors controlling floral trichome development in Aristolochia (Aristolochiaceae). J. Exp. Bot. 2023, 74, 6588–6607. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Mirlohi, S.; Li, X.; He, Y. Identification of functional single-nucleotide polymorphisms affecting leaf hair number in Brassica rapa. Plant Physiol. 2018, 177, 490–503. [Google Scholar] [CrossRef]
- Xu, B.; Zhang, J.; Shi, Y.; Dai, F.; Jiang, T.; Xuan, L.; He, Y.; Zhang, Z.; Deng, J.; Zhang, T.; et al. GoSTR, a negative modulator of stem trichome formation in cotton. Plant J. 2023, 116, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.Y.; Miao, H.; Ding, L.H.; Wehner, T.C.; Liu, P.N.; Wang, Y.; Zhang, S.P.; Gu, X.F. A New glabrous gene (csgl3) identified in trichome development in cucumber (Cucumis sativus L.). PLoS ONE 2016, 11, e0148422. [Google Scholar] [CrossRef]
- Yuan, R.; Cao, Y.; Li, T.; Yang, F.; Yu, L.; Qin, Y.; Du, X.; Liu, F.; Ding, M.; Jiang, Y.; et al. Differentiation in the genetic basis of stem trichome development between cultivated tetraploid cotton species. BMC Plant Biol. 2021, 21, 115. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chu, L.; Liu, X.; Zhang, N.; Xu, Y.; Karikari, B.; Wang, Y.; Chang, F.; Liu, Z.; Tan, L.; et al. Genetic architecture and candidate genes for pubescence length and density and its relationship with resistance to common cutworm in soybean. Front. Plant Sci. 2022, 12, 771850. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Zhang, J.; Gong, D.; Zhang, Z.; Yu, Y.; Luo, G.; Somta, P.; Hu, Z.; Wang, S.; Yuan, X.; et al. Genomic analyses of rice bean landraces reveal adaptation and yield related loci to accelerate breeding. Nat. Commun. 2022, 13, 5707. [Google Scholar] [CrossRef] [PubMed]
- Pootakham, W.; Nawae, W.; Naktang, C.; Sonthirod, C.; Yoocha, T.; Kongkachana, W.; Sangsrakru, D.; Jomchai, N.; U-Thoomporn, S.; Somta, P.; et al. A chromosome-scale assembly of the black gram (Vigna mungo) genome. Mol. Ecol. Resour. 2021, 21, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Jegadeesan, S.; Raizada, A.; Dhanasekar, P.; Suprasanna, P. Draft genome sequence of the pulse crop blackgram [Vigna mungo (L.) Hepper] reveals potential R-genes. Sci. Rep. 2021, 11, 11247. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Holt, K.E.; Teo, Y.Y.; Li, H.; Nair, S.; Dougan, G.; Wain, J.; Parkhill, J. Detecting SNPs and estimating allele frequencies in clonal bacterial populations by sequencing pooled DNA. Bioinformatics 2009, 25, 2074–2075. [Google Scholar] [CrossRef]
- Huang, X.; Feng, Q.; Qian, Q.; Zhao, Q.; Wang, L.; Wang, A.; Guan, J.; Fan, D.; Weng, Q.; Huang, T.; et al. Highthroughput genotyping by whole-genome resequencing. Genome Res. 2009, 19, 1068–1076. [Google Scholar] [CrossRef]
- Kulwal, P.L. Trait Mapping Approaches Through Linkage Mapping in Plants. Adv Biochem Eng Biotechnol 2018, 164, 53–82. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Kundu, A.; Patel, A.; Pal, A. Defining reference genes for qPCR normalization to study biotic and abiotic stress responses in Vigna mungo. Plant Cell Rep. 2013, 32, 1647–1658. [Google Scholar] [CrossRef]
- Yang, C.; Li, H.; Zhang, J.; Luo, Z.; Gong, P.; Zhang, C.; Li, J.; Wang, T.; Zhang, Y.; Lu, Y.; et al. A regulatory gene induces trichome formation and embryo lethality in tomato. Proc. Natl. Acad. Sci. USA 2011, 8, 11836–11841. [Google Scholar] [CrossRef]
- Chen, J.J.; Mbogning, J.; Hancock, M.A.; Majdpour, D.; Madhok, M.; Nassour, H.; Dallagnol, J.C.; Pagé, V.; Chatenet, D.; Tanny, J.C. Spt5 Phosphorylation and the Rtf1 plus3 domain promote Rtf1 function through distinct mechanisms. Mol. Cell Biol. 2020, 40, e00150-20. [Google Scholar] [CrossRef] [PubMed]
- Mbogning, J.; Nagy, S.; Pagé, V.; Schwer, B.; Shuman, S.; Fisher, R.P.; Tanny, J.C. The PAF complex and Prf1/Rtf1 delineate distinct Cdk9-dependent pathways regulating transcription elongation in fission yeast. PLoS Genet. 2013, 9, e1004029. [Google Scholar] [CrossRef] [PubMed]
- Koyauchi, T.; Niida, H.; Motegi, A.; Sakai, S.; Uchida, C.; Ohhata, T.; Iijima, K.; Yokoyama, A.; Suda, T.; Kitagawa, M. Chromatin-remodeling factor BAZ1A/ACF1 targets UV damage sites in an MLL1-dependent manner to facilitate nucleotide excision repair. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119332. [Google Scholar] [CrossRef] [PubMed]
- Exner, V.; Gruissem, W.; Hennig, L. Control of trichome branching by chromatin assembly factor-1. BMC Plant. Biol. 2008, 8, 54. [Google Scholar] [CrossRef]
- Fonseca, R.; Capel, C.; Yuste-Lisbona, F.J.; Quispe, J.L.; Gómez-Martín, C.; Lebrón, R.; Hackenberg, M.; Oliver, J.L.; Angosto, T.; Lozano, R.; et al. Functional characterization of the tomato HAIRPLUS gene reveals the implication of the epigenome in the control of glandular trichome formation. Hortic. Res. 2022, 9, uhab015. [Google Scholar] [CrossRef] [PubMed]
- Kotak, J.; Saisana, M.; Gegas, V.; Pechlivani, N.; Kaldis, A.; Papoutsoglou, P.; Makris, A.; Burns, J.; Kendig, A.L.; Sheikh, M.; et al. The histone acetyltransferase GCN5 and the transcriptional coactivator ADA2b affect leaf development and trichome morphogenesis in Arabidopsis. Planta 2018, 248, 613–628. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Linkages | Total Bin Marker | SNP Numbers | Total Distance (cM) | Average Distance (cM) | Max Gap (cM) | Gaps < 5cM (%) |
|---|---|---|---|---|---|---|
| LG2373 | 835 | 34,278 | 116.59 | 0.14 | 1.3 | 100% |
| LG2711 | 446 | 14,688 | 145.62 | 0.33 | 3.22 | 100% |
| LG4236 | 480 | 17,664 | 140.77 | 0.29 | 4.05 | 100% |
| LG4343 | 545 | 16,824 | 175.06 | 0.32 | 2.65 | 100% |
| LG5881 | 559 | 19,318 | 163.13 | 0.29 | 2.39 | 100% |
| LG5891 | 479 | 15,862 | 153.28 | 0.32 | 3.22 | 100% |
| LG10972 | 293 | 10,189 | 119.09 | 0.41 | 2.65 | 100% |
| LG11446 | 345 | 10,877 | 122.16 | 0.36 | 4.31 | 100% |
| LG11447 | 470 | 15,522 | 142.25 | 0.3 | 3.53 | 100% |
| LG11448 | 301 | 7296 | 150 | 0.5 | 4.36 | 100% |
| LG11449 | 981 | 21,940 | 183.49 | 0.19 | 2.13 | 100% |
| Gene | Gene Location | Annotation |
|---|---|---|
| Scaffold_9372_HRSCAF_11447.163 | Chr6: 834,818–839,129 | stAR-related lipid transfer protein 7 |
| Scaffold_9372_HRSCAF_11447.164 | Chr6: 842,193–847,000 | RNA polymerase-associated protein Rtf1 |
| Scaffold_9372_HRSCAF_11447.165 | Chr6: 848,491–849,285 | CDI (cadmium 2+ induced) |
| Scaffold_9372_HRSCAF_11447.166 | Chr6: 858,351–865,191 | Bromodomain adjacent to zinc-finger domain protein 1A |
| Scaffold_9372_HRSCAF_11447.167 | Chr6: 867,211–869,064 | Histone demethylase JARID1 |
| Scaffold_9372_HRSCAF_11447.168 | Chr6: 870,460–872,091 | RGF1 INDUCIBLE TRANSCRIPTION FACTOR 1 |
| Gene_Id | Position | P1 (Trichome) | P2 (Glabrous) | Location/Effect |
|---|---|---|---|---|
| Scaffold_9372_HRSCAF_11447.163 | 836,049 | G | GT | Intron |
| 838,193 | A | AT | Intron | |
| 839,519 | C | CAAAAGAAAAG | Downstream | |
| 839,654 | AT | A | Downstream | |
| Scaffold_9372_HRSCAF_11447.164 | 842,357 | TCCG | T | Codon_insertion |
| 843,363 | AAAG | A | Intron | |
| Scaffold_9372_HRSCAF_11447.165 | 848,154 | A | ACAGAG | Upstream |
| 848,319 | T | TA | Upstream | |
| 849,984 | AATT | A | Downstream | |
| Scaffold_9372_HRSCAF_11447.166 | 855,253 | T | TA | Upstream |
| 855,365 | A | ATTTT | Upstream | |
| Scaffold_9372_HRSCAF_11447.168 | 873,299 | G | GT | Downstream |
| 873,465 | T | TA | Downstream | |
| 873,655 | T | TATTATC | Downstream |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, D.; Li, J.; Wang, S.; Sha, A.; Wang, L. Mapping and Detection of Genes Related to Trichome Development in Black Gram (Vigna mungo (L.) Hepper). Genes 2024, 15, 308. https://doi.org/10.3390/genes15030308
Gong D, Li J, Wang S, Sha A, Wang L. Mapping and Detection of Genes Related to Trichome Development in Black Gram (Vigna mungo (L.) Hepper). Genes. 2024; 15(3):308. https://doi.org/10.3390/genes15030308
Chicago/Turabian StyleGong, Dan, Jianling Li, Suhua Wang, Aihua Sha, and Lixia Wang. 2024. "Mapping and Detection of Genes Related to Trichome Development in Black Gram (Vigna mungo (L.) Hepper)" Genes 15, no. 3: 308. https://doi.org/10.3390/genes15030308
APA StyleGong, D., Li, J., Wang, S., Sha, A., & Wang, L. (2024). Mapping and Detection of Genes Related to Trichome Development in Black Gram (Vigna mungo (L.) Hepper). Genes, 15(3), 308. https://doi.org/10.3390/genes15030308