

Genome-Wide Mapping of Quantitative Trait Loci for Yield-Attributing Traits of Peanut
 ,
,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Phenotyping RIL Population for Yield and Quality Traits
2.1.1. Phenotyping for Yield-Related Traits
2.1.2. Phenotyping for Physiological Traits
2.1.3. Phenotyping for Oil Content, Fatty Acids, and Protein Content
2.2. Phenotypic Analysis
2.3. DNA Isolation and Sequencing
2.4. SNP Calling and Filtering
2.5. Construction of Genetic Linkage Map and QTL Analysis
2.6. Identification of Candidate Genes from Identified QTL Regions and Expression Analysis
2.7. Identification of Epistatic (Q × Q) Effect
3. Results
3.1. Phenotypic Data Analysis
3.2. Identification of Marker Polymorphism and Genotyping
3.3. Construction of Genetic Linkage Map
3.4. QTLs for Yield and Quality Traits
3.4.1. QTLs for Yield-Related and Physiological Traits
3.4.2. QTLs for Oil Content, Fatty Acids, and Protein Content
3.5. Epistatic (QTL × QTL) Interaction for Yield- and Quality-Related Traits
3.5.1. Digenic Interaction
3.5.2. Trigenic Interaction
3.6. Identification of Candidate Genomic Regions for HSW and SP
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Toomer, O.T. Nutritional chemistry of the peanut (Arachis hypogaea). Crit. Rev. Food Sci. Nutr. 2018, 58, 3042–3053. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.P.; Dean, L.L. Chapter 11—Peanut composition, flavor and nutrition A2. In Peanuts; Stalker, H.T., Wilson, R.F., Eds.; AOCS Press: Urbana, IL, USA, 2016; pp. 289–345. [Google Scholar]
- Nigam, S.N. Groundnut at a Glance; ICRISAT: Hyderabad, India, 2014; p. 121. [Google Scholar]
- FAOSTAT. 2021. Available online: https://www.fao.org/faostat/en/#data/QCL (accessed on 10 January 2023).
- Chen, Y.; Ren, X.; Zheng, Y.; Zhou, X.; Huang, L.; Yan, L.; Jiao, Y.; Chen, W.; Huang, S.; Wan, L.; et al. Genetic mapping of yield traits using RIL population derived from Fuchuan Dahuasheng and ICG6375 of peanut (Arachis hypogaea L.). Mol. Breed. 2017, 37, 17. [Google Scholar] [CrossRef] [PubMed]
- Denwar, N.N.; Simpson, C.E.; Starr, J.L.; Wheeler, T.A.; Burow, M.D. Evaluation and selection of interspecific lines of groundnut (Arachis hypogaea L.) for resistance to leaf spot disease and for yield improvement. Plants 2021, 10, 873. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Liu, H.; Qin, L.; Qi, F.; Sun, Z.; Wu, J.; Dong, W.; Huang, B.; Zhang, X. Identification of QTL for kernel weight and size and analysis of the pentatricopeptide repeat (PPR) gene family in cultivated peanut (Arachis hypogaea L.). BMC Genom. 2023, 24, 495. [Google Scholar] [CrossRef]
- Shanthala, J.; Gazala Parveen, S.; Jambagi, B.K.P. Genomic-Assisted Breeding for Enhanced Harvestable (Pod) and Consumable (Seed) Product, Yield Productivity in Groundnut (Arachis hypogaea L.). In Accelerated Plant Breeding, Volume 4; Oil Crops: Wuhan, China, 2022; pp. 181–237. [Google Scholar]
- Bertioli, D.J.; Cannon, S.B.; Froenicke, L.; Huang, G.; Farmer, A.D.; Cannon, E.K.; Liu, X.; Gao, D.; Clevenger, J.; Dash, S.; et al. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat. Genet. 2016, 48, 438–446. [Google Scholar] [CrossRef]
- Kochert, G.; Halward, T.; Branch, W.D.; Simpson, C.E. RFLP variability in peanut (Arachis hypogaea L.) cultivars and wild species. Theor. Appl. Genet. 1991, 81, 565–570. [Google Scholar] [CrossRef]
- Nielen, S.; Vidigal, B.S.; Leal-Bertioli, S.C.; Ratnaparkhe, M.; Paterson, A.H.; Garsmeur, O.; D’Hont, A.; Guimaraes, P.M.; Bertioli, D.J. Matita, a new retroelement from peanut: Characterization and evolutionary context in the light of the Arachis A–B genome divergence. Mol. Genet. Genom. 2012, 287, 21–38. [Google Scholar] [CrossRef]
- Guimarães, P.M.; Brasileiro, A.C.M.; Mehta, A.; Araujo, A.C.G. Functional genomics in peanut wild relatives. In The Peanut Genome; Springer: Berlin/Heidelberg, Germany, 2017; pp. 149–164. [Google Scholar]
- Liang, Y.; Baring, M.R.; Septiningsih, E.M. Mapping of quantitative trait loci for yield and grade related traits in peanut (Arachis hypogaea L.) using high-resolution SNP markers. Plant Breed. Biotechnol. 2018, 6, 454–462. [Google Scholar] [CrossRef]
- Wang, L.; Yang, X.; Cui, S.; Mu, G.; Sun, X.; Liu, L.; Li, Z. QTL mapping and QTL× environment interaction analysis of multi-seed pod in cultivated peanut (Arachis hypogaea L.). Crop J. 2019, 7, 249–260. [Google Scholar] [CrossRef]
- Chu, Y.; Chee, P.; Culbreath, A.; Isleib, T.G.; Holbrook, C.C.; Ozias-Akins, P. Major QTLs for resistance to early and late leaf spot diseases are identified on chromosomes 3 and 5 in peanut (Arachis hypogaea). Front. Plant Sci. 2019, 10, 883. [Google Scholar] [CrossRef]
- Selvaraj, M.G.; Narayana, M.; Schubert, A.M.; Ayers, J.L.; Baring, M.R.; Burow, M.D. Identification of QTLs for pod and kernel traits in cultivated peanut by bulked segregant analysis. Electron. J. Biotechnol. 2009, 12, 3–4. [Google Scholar] [CrossRef]
- Hong, Y.B.; Liang, X.Q.; Chen, X.P.; Liu, H.Y.; Zhou, G.Y.; Li, S.X.; Wen, S.J. Construction of genetic linkage map based on SSR markers in peanut (Arachis hypogaea L.). Agric. Sci. China 2008, 7, 915–921. [Google Scholar] [CrossRef]
- Wang, H.; Penmetsa, R.V.; Yuan, M.; Gong, L.; Zhao, Y.; Guo, B.; Farmer, A.D.; Rosen, B.D.; Gao, J.; Isobe, S.; et al. Development and characterization of BAC-end sequence derived SSRs, and their incorporation into a new higher density genetic map for cultivated peanut (Arachis hypogaea L.). BMC Plant Biol. 2012, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.D.; Singh, A.K. Marker-Assisted Plant Breeding: Principles and Practices; Springer: Berlin/Heidelberg, Germany, 2015; pp. 47–75. [Google Scholar]
- Liao, P.Y.; Lee, K.H. From SNPs to functional polymorphism: The insight into biotechnology applications. Biochem. Eng. J. 2010, 49, 149–158. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, L.; Ren, M.; Xiang, C.; Tang, X.; Xia, Y.; Song, D.; Li, F. Genome-Wide Association Studies Revealed the Genetic Loci and Candidate Genes of Pod-Related Traits in Peanut (Arachis hypogaea L.). Agronomy 2023, 13, 1863. [Google Scholar] [CrossRef]
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H.; et al. SLAF-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 2013, 8, 58700. [Google Scholar] [CrossRef]
- Wang, Z.; Huai, D.; Zhang, Z.; Cheng, K.; Kang, Y.; Wan, L.; Yan, L.; Jiang, H.; Lei, Y.; Liao, B. Development of a high-density genetic map based on specific length amplified fragment sequencing and its application in quantitative trait loci analysis for yield-related traits in cultivated peanut. Front. Plant Sci. 2018, 9, 827. [Google Scholar] [CrossRef]
- Gangurde, S.S.; Khan, A.W.; Janila, P.; Variath, M.T.; Manohar, S.S.; Singam, P.; Chitikineni, A.; Varshney, R.K.; Pandey, M.K. Whole-genome sequencing based discovery of candidate genes and diagnostic markers for seed weight in groundnut. Plant Genome 2022, 16, e20265. [Google Scholar] [CrossRef]
- Luo, H.; Pandey, M.K.; Khan, A.W.; Guo, J.; Wu, B.; Cai, Y.; Huang, L.; Zhou, X.; Chen, Y.; Chen, W.; et al. Discovery of genomic regions and candidate genes controlling shelling percentage using QTL-seq approach in cultivated peanut (Arachis hypogaea L.). Plant Biotechnol. J. 2019, 17, 1248–1260. [Google Scholar] [CrossRef]
- Sharma, V.; Gangurde, S.S.; Nayak, S.N.; Gowda, A.S.; Sukanth, B.S.; Mahadevaiah, S.S.; Manohar, S.S.; Choudhary, R.S.; Anitha, T.; Malavalli, S.S.; et al. Genetic mapping identified three hotspot genomic regions and candidate genes controlling heat tolerance-related traits in groundnut. Front. Plant Sci. 2023, 14, 1182867. [Google Scholar] [CrossRef]
- Liang, W.A.N.G.; Yang, X.L.; Cui, S.L.; Wang, J.H.; Hou, M.Y.; Mu, G.J.; Li, Z.C.; Liu, L.F. Identification of main effect and epistatic QTLs controlling initial flowering date in cultivated peanut (Arachis hypogaea L.). J. Integr. Agric. 2020, 19, 2383–2393. [Google Scholar]
- Monnahan, P.J.; Kelly, J.K. Epistasis is a major determinant of the additive genetic variance in Mimulus guttatus. PLoS Genet. 2015, 11, e1005201. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Cui, R.; Chavarro, C.; Tseng, Y.C.; Zhou, H.; Peng, Z.; Chu, Y.; Yang, X.; Lopez, Y.; Tillman, B.; et al. Mapping quantitative trait loci (QTLs) and estimating the epistasis controlling stem rot resistance in cultivated peanut (Arachis hypogaea). Theor. App. Genet. 2020, 133, 1201–1212. [Google Scholar] [CrossRef] [PubMed]
- Blum, A. Improving wheat grain filling under stress by stem reserve mobilisation. Euphytica 1998, 100, 77–83. [Google Scholar] [CrossRef]
- Reddy, G.K. Genetics of SCMR and SLA, the Traits Related to Drought Tolerance in Groundnut (Arachis hypogaea L.). Ph.D. Thesis, University of Agricultural Sciences, Bangalore, India, 2008. [Google Scholar]
- Puppala, N.; Mura, J.D.; Vadez, V.; Paspuleti, J.; Pandey, M.K.; Varshney, R.K. Integrated Agronomy, Physiology, and Plant Breeding Approaches to Improve Drought Tolerance Phenotyping in Peanut; APRES, Inc.: Williamsburg, VA, USA, 2019; p. 157. [Google Scholar]
- Hsi, D.C. Regisration of New Mexico Valencia C. Crop Sci. 1980, 20, 113–114. [Google Scholar] [CrossRef]
- Cemal, K.U.R.T.; Bakal, H.; Gulluoglu, L.; Arioglu, H. The effect of twin row planting pattern and plant population on yield and yield components of peanut (Arachis hypogaea L.) at main crop planting in Cukurova region of Turkey. Turk. J. Field Crops 2017, 22, 24–31. [Google Scholar]
- Nageswara Rao, R.C.; Talwar, H.S.; Wright, G.C. Rapid assessment of specific leaf area and leaf nitrogen in peanut (Arachis hypogaea L.) using a chlorophyll meter. J. Agron. Crop Sci. 2001, 186, 175–182. [Google Scholar] [CrossRef]
- Sundaram, J.; Kandala, C.V.; Butts, C.L. Application of near infrared spectroscopy to peanut grading and quality analysis: Overview. Sens. Instrum. Food Qual. Saf. 2009, 3, 156–164. [Google Scholar] [CrossRef]
- Popat, R.; Patel, R.; Parmar, D. Variability: Genetic Variability Analysis for Plant Breeding Research. R Package Version 0.1.0. 2020. Available online: https://CRAN.R-project.org/package=variability (accessed on 13 June 2021).
- Pandey, M.K.; Gangurde, S.S.; Sharma, V.; Pattanashetti, S.K.; Naidu, G.K.; Faye, I.; Hamidou, F.; Desmae, H.; Kane, N.A.; Yuan, M.; et al. Improved genetic map identified major QTLs for drought tolerance-and iron deficiency tolerance-related traits in groundnut. Genes 2021, 12, 37. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, S.A.; Kawamoto, K.; Buckler, E.S.; Mitchel, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Clarke, W.E.; Higgins, E.E.; Plieske, J.; Wieseke, R.; Sidebottom, C.; Khedikar, Y.; Batley, J.; Edwards, D.; Meng, J.; Li, R.; et al. A high-density SNP genotyping array for Brassica napus and its ancestral diploid species based on optimised selection of single-locus markers in the allotetraploid genome. Theor. Appl. Genet. 2016, 129, 1887–1899. [Google Scholar] [CrossRef]
- Van Ooijen, J.W. JoinMap4, Software for the Calculation of Genetic Linkage Maps in Experimental Populations; Kyazma BV: Wageningen, The Netherlands, 2016. [Google Scholar]
- Ouellette, L.A. LinkageMapView-Rendering high resolution linkage and QTL maps. Bioinformatics 2018, 34, 306–307. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Bastern, J.; Zeng, Z.B. Windows QTL Cartographer 2.5; Department of Statistics, North Carolina State University: Raleigh, NC, USA, 2011. [Google Scholar]
- Sinha, P.; Bajaj, P.; Pazhamala, L.T.; Nayak, S.N.; Pandey, M.K.; Chitikineni, A.; Huai, D.; Khan, A.W.; Desai, A.; Jiang, H.; et al. Arachis hypogaea gene expression atlas for fastigiata subspecies of cultivated groundnut to accelerate functional and translational genomics applications. Plant Biotechnol. J. 2020, 18, 2187–2200. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, H.; Zhang, L.; Meng, L. Users’ Manual of QTL Ici-Mapping Version 3.3; Chinese Academy of Agricultural Sciences (CAAS): Beijing, China, 2012. [Google Scholar]
- Miao, P.; Meng, X.; Li, Z.; Sun, S.; Chen, C.Y.; Yang, X. Mapping Quantitative Trait Loci (QTLs) for hundred-pod and hundred-seed weight under seven environments in a recombinant inbred line population of cultivated peanut (Arachis hypogaea L.). Genes 2023, 14, 1792. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zheng, H.; Jin, L.; Xing, L.; Zou, J.; Zhang, L.; Liu, C.; Chu, J.; Xu, M.; Wang, L. miR169o and ZmNF-YA13 act in concert to coordinate the expression of ZmYUC1 that determines seed size and weight in maize kernels. New Phytol. 2022, 235, 2270–2284. [Google Scholar] [CrossRef]
- Luo, S.; Jia, J.; Liu, R.; Wei, R.; Guo, Z.; Cai, Z.; Chen, B.; Liang, F.; Xia, Q.; Nian, H.; et al. Identification of major QTLs for soybean seed size and seed weight traits using a RIL population in different environments. Front. Plant Sci. 2023, 13, 1094112. [Google Scholar] [CrossRef]
- Sun, C.; Wang, Y.; Yang, X.; Tang, L.; Wan, C.; Liu, J.; Chen, C.; Zhang, H.; He, C.; Liu, C.; et al. MATE transporter GFD1 cooperates with sugar transporters, mediates carbohydrate partitioning and controls grain-filling duration, grain size and number in rice. Plant Biotechnol. J. 2023, 21, 621–634. [Google Scholar] [CrossRef]
- Huang, L.; He, H.; Chen, W.; Ren, X.; Chen, Y.; Zhou, X.; Xia, Y.; Wang, X.; Jiang, X.; Liao, B.; et al. Quantitative trait locus analysis of agronomic and quality-related traits in cultivated peanut (Arachis hypogaea L.). Theor. App. Genet. 2015, 128, 1103–1115. [Google Scholar] [CrossRef]
- Luo, H.; Xu, Z.; Li, Z.; Li, X.; Lv, J.; Ren, X.; Huang, L.; Zhou, X.; Chen, Y.; Yu, J.; et al. Development of SSR markers and identification of major quantitative trait loci controlling shelling percentage in cultivated peanut (Arachis hypogaea L.). Theor. Appl. Genet. 2017, 130, 1635–1648. [Google Scholar] [CrossRef]
- Ajay, B.C.; Bera, S.K.; Singh, A.L.; Kumar, N.; Gangadhar, K.; Kona, P. Evaluation of genotype× environment interaction and yield stability analysis in peanut under phosphorus stress condition using stability parameters of AMMI model. Agric. Res. 2020, 9, 477–486. [Google Scholar] [CrossRef]
- Varshney, R.K.; Pandey, M.K.; Janila, P.; Nigam, S.N.; Sudini, H.; Gowda, M.V.C.; Sriswathi, M.; Radhakrishnan, T.; Manohar, S.S.; Nagesh, P. Marker-assisted introgression of a QTL region to improve rust resistance in three elite and popular varieties of peanut (Arachis hypogaea L.). Theor. Appl. Genet. 2014, 127, 1771–1781. [Google Scholar] [CrossRef]
- Pasupuleti, J.; Pandey, M.K.; Manohar, S.S.; Variath, M.T.; Nallathambi, P.; Nadaf, H.L.; Sudini, H.; Varshney, R.K. Foliar fungal disease-resistant introgression lines of groundnut (Arachis hypogaea L.) record higher pod and haulm yield in multilocation testing. Plant Breed. 2016, 135, 355–366. [Google Scholar] [CrossRef]
- Ramakrishnan, P.; Manivannan, N.; Mothilal, A.; Mahaingam, L.; Prabhu, R.; Gopikrishnan, P. Marker assisted introgression of QTL region to improve late leaf spot and rust resistance in elite and popular variety of groundnut (Arachis hypogaea L.) cv TMV 2. Australas. Plant Dis. 2020, 49, 505–513. [Google Scholar] [CrossRef]
- Pandey, M.K.; Agarwal, G.; Kale, S.M.; Clevenger, J.; Nayak, S.N.; Sriswathi, M.; Chitikineni, A.; Chavarro, C.; Chen, X.; Upadhyaya, H.D.; et al. Development and evaluation of a high-density genotyping ‘Axiom_Arachis’ array with 58 K SNPs for accelerating genetics and breeding in groundnut. Sci. Rep. 2017, 7, 40577. [Google Scholar] [CrossRef]
- Gangurde, S.S.; Pasupuleti, J.; Parmar, S.; Variath, M.T.; Bomireddy, D.; Manohar, S.S.; Varshney, R.K.; Singam, P.; Guo, B.; Pandey, M.K. Genetic mapping identifies genomic regions and candidate genes for seed weight and shelling percentage in groundnut. Front. Genet. 2023, 14, 1128182. [Google Scholar] [CrossRef]
- Yang, Y.; Su, Q.; Li, Y.; Cheng, Z.; Song, Y.; Jin, X.; Wang, J. Fine mapping of a major QTL qHYF_B06 for peanut yield. Crop J. 2023, 11, 1533–1540. [Google Scholar] [CrossRef]
- Luo, H.; Guo, J.; Ren, X.; Chen, W.; Huang, L.; Zhou, X.; Chen, Y.; Liu, N.; Xiong, F.; Lei, Y.; et al. Chromosomes A07 and A05 associated with stable and major QTLs for pod weight and size in cultivated peanut (Arachis hypogaea L.). Theor. Appl. Genet. 2018, 131, 267–282. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, N.; Huang, L.; Chen, Y.; Guo, J.; Yu, B.; Luo, H.; Zhou, X.; Huai, D.; Chen, W.; et al. Stable major QTL on chromosomes A07 and A08 increase shelling percentage in peanut (Arachis hypogaea L.). Crop J. 2022, 10, 820–829. [Google Scholar] [CrossRef]
- Zhang, S.; Hu, X.; Miao, H.; Chu, Y.; Cui, F.; Yang, W.; Wang, C.; Shen, Y.; Xu, T.; Zhao, L.; et al. QTL identification for seed weight and size based on a high-density SLAF-seq genetic map in peanut (Arachis hypogaea L.). BMC Plant Biol. 2019, 19, 537. [Google Scholar] [CrossRef]
- Hu, Z.; He, H.; Zhang, S.; Sun, F.; Xin, X.; Wang, W.; Qian, X.; Yang, J.; Luo, X. A Kelch motif-containing serine/threonine protein phosphatase determines the large grain QTL trait in rice. J. Integr. Plant Biol. 2012, 54, 979–990. [Google Scholar] [CrossRef]
- Jiang, H.; Li, Y.; Qin, H.; Li, Y.; Qi, H.; Li, C.; Wang, N.; Li, R.; Zhao, Y.; Huang, S.; et al. Identification of major QTLs associated with first pod height and candidate gene mining in soybean. Front. Plant Sci. 2018, 9, 1280. [Google Scholar] [CrossRef]
- Jia, H.; Li, M.; Li, W.; Liu, L.; Jian, Y.; Yang, Z.; Shen, X.; Ning, Q.; Du, Y.; Zhao, R.; et al. A serine/threonine protein kinase encoding gene KERNEL NUMBER PER ROW6 regulates maize grain yield. Nat. Commun. 2020, 11, 988. [Google Scholar] [CrossRef]
- Wu, Y.; Sun, Z.; Qi, F.; Tian, M.; Wang, J.; Zhao, R.; Wang, X.; Wu, X.; Shi, X.; Liu, H.; et al. Comparative transcriptomics analysis of developing peanut (Arachis hypogaea L.) pods reveals candidate genes affecting peanut seed size. Front. Plant Sci. 2022, 13, 958808. [Google Scholar] [CrossRef]
- Jain, M.; Pathak, B.P.; Harmon, A.C.; Tillman, B.L.; Gallo, M. Calcium dependent protein kinase (CDPK) expression during fruit development in cultivated peanut (Arachis hypogaea) under Ca2+-sufficient and-deficient growth regimens. J. Plant Physiol. 2011, 168, 2272–2277. [Google Scholar] [CrossRef]
- Bi, Y.P.; Liu, W.; Xia, H.; Su, L.; Zhao, C.Z.; Wan, S.B.; Wang, X.J. EST sequencing and gene expression profiling of cultivated peanut (Arachis hypogaea L.). Genome 2010, 53, 832–839. [Google Scholar] [CrossRef]
- Gao, M.J.; Lydiate, D.J.; Li, X.; Lui, H.; Gjetvaj, B.; Hegedus, D.D.; Rozwadowski, K. Repression of seed maturation genes by a trihelix transcriptional repressor in Arabidopsis seedlings. Plant Cell 2009, 21, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Sun, Q.; Kong, Y. Genome-wide mining seed-specific candidate genes from peanut for promoter cloning. PLoS ONE 2019, 14, 0214025. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Gu, Q.; Zhang, J.; Sun, L.; Kuppu, S.; Zhang, Y.; Burow, M.; Payton, P.; Blumwald, E.; Zhang, H. Regulated expression of an isopentenyltransferase gene (IPT) in peanut significantly improves drought tolerance and increases yield under field conditions. Plant Cell Physiol. 2011, 52, 1904–1914. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Zhou, D.; Shi, X.; Ren, J.; Zhang, H.; Zhong, C.; Kang, S.; Zhao, X.; Yu, H.; Wang, C. The determination of peanut (Arachis hypogaea L.) pod-sizes during the rapid-growth stage by phytohormones. BMC Plant Biol. 2023, 23, 371. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, P.; Xia, H.; Zhao, C.; Hou, L.; Li, C.; Gao, C.; Zhao, S.; Wang, X. Comparative transcriptome analysis of basal and zygote-located tip regions of peanut ovaries provides insight into the mechanism of light regulation in peanut embryo and pod development. BMC Genom. 2016, 17, 606. [Google Scholar]
- Yang, S.; Wang, J.; Tang, Z.; Guo, F.; Zhang, Y.; Zhang, J.; Meng, J.; Zheng, L.; Wan, S.; Li, X. Transcriptome of peanut kernel and shell reveals the mechanism of calcium on peanut pod development. Sci. Rep. 2020, 10, 15723. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.; Hou, X.L.; Xing, G.M.; Liu, J.X.; Duan, A.Q.; Xu, Z.S.; Li, M.Y.; Zhuang, J.; Xiong, A.S. Advances in AP2/ERF super-family transcription factors in plant. Crit. Rev. Biotechnol. 2020, 40, 750–776. [Google Scholar] [CrossRef]
- Cui, M.; Haider, M.S.; Chai, P.; Guo, J.; Du, P.; Li, H.; Dong, W.; Huang, B.; Zheng, Z.; Shi, L.; et al. Genome-wide identification and expression analysis of AP2/ERF transcription factor related to drought stress in cultivated peanut (Arachis hypogaea L.). Front. Genet. 2021, 12, 750761. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhang, X.; Song, X.; Yang, J.; Pang, Y. Genome-wide identification and characterization of APETALA2/ethylene-responsive element binding factor superfamily genes in soybean seed development. Front. Plant Sci. 2020, 11, 566647. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Nakazawa, M.; Shibata, K.; Yokota, T.; Ishikawa, A.; Suzuki, K.; Kawashima, M.; Ichikawa, T.; Shimada, H.; Matsui, M. shk1-D, a dwarf Arabidopsis mutant caused by activation of the CYP72C1 gene, has altered brassinosteroid levels. Plant J. 2005, 42, 13–22. [Google Scholar] [CrossRef]
- Yoo, K.S.; Ok, S.H.; Jeong, B.C.; Jung, K.W.; Cui, M.H.; Hyoung, S.; Lee, M.R.; Song, H.K.; Shin, J.S. Single cystathionine β-synthase domain–containing proteins modulate development by regulating the thioredoxin system in Arabidopsis. Plant Cell 2011, 23, 3577–3594. [Google Scholar] [CrossRef]
- Hao, Q.; Shang, W.; Zhang, C.; Chen, H.; Chen, L.; Yuan, S.; Chen, S.; Zhang, X.; Zhou, X. Identification and comparative analysis of CBS domain-containing proteins in soybean (Glycine max) and the primary function of GmCBS21 in enhanced tolerance to low nitrogen stress. Int. J. Mol. Sci. 2016, 17, 620. [Google Scholar] [CrossRef]
- Henriksson, M.; Lüscher, B. Proteins of the MYC network: Essential regulators of cell growth and differentiation. Adv. Cancer Res. 1996, 68, 109–182. [Google Scholar]
- Zhang, X.; Zhang, J.; He, X.; Wang, Y.; Ma, X.; Yin, D. Genome-wide association study of major agronomic traits related to domestication in peanut. Front. Plant Sci. 2017, 8, 1611. [Google Scholar] [CrossRef] [PubMed]
- Heim, M.A.; Jakoby, M.; Werber, M.; Martin, C.; Weisshaar, B.; Bailey, P.C. The basic helix–loop–helix transcription factor family in plants: A genome-wide study of protein structure and functional diversity. Mol. Biol. Evol. 2003, 20, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Kondou, Y.; Nakazawa, M.; Kawashima, M.; Ichikawa, T.; Yoshizumi, T.; Suzuki, K.; Ishikawa, A.; Koshi, T.; Matsui, R.; Muto, S.; et al. RETARDED GROWTH of EMBRYO1, a new basic helix-loop-helix protein, expresses in endosperm to control embryo growth. Plant Physiol. 2008, 147, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Fu, Y.; Lee, Y.R.J.; Chern, M.; Li, M.; Cheng, M.; Dong, H.; Yuan, Z.; Gui, L.; Yin, J.; et al. The PGS1 basic helix-loop-helix protein regulates Fl3 to impact seed growth and grain yield in cereals. J. Plant Biotechnol. 2022, 20, 1311–1326. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, Q.; Yan, C.; Sun, Q.; Li, C.; Yuan, C.; Mou, Y.; Shan, S. The bHLH transcription factor AhbHLH121 improves salt tolerance in peanut. Int. J. Biol. Macromol. 2023, 256, 128492. [Google Scholar] [CrossRef] [PubMed]
- Jannink, J.L. Identifying quantitative trait locus by genetic background interactions in association studies. Genetics 2007, 176, 553–561. [Google Scholar] [CrossRef][Green Version]
- Isobe, S.; Nakaya, A.; Tabata, S. Genotype matrix mapping: Searching for quantitative trait loci interactions in genetic variation in complex traits. DNA Res. 2007, 14, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, R. Multiparental populations in line development: Genetic gain, diversity, and practical limitations. Crop Sci. 2021, 61, 4139–4150. [Google Scholar] [CrossRef]
- Zhang, S.; Hu, X.; Wang, F.; Miao, H.; Chu, Y.; Yang, W.; Cui, F.; Xu, S.; Guo, J.; Yu, H.; et al. Genetic dissection of additive and epistatic quantitative trait loci controlling pod number per plant in peanut (Arachis hypogaea L.). Euphytica 2023, 219, 31. [Google Scholar] [CrossRef]
- Zhang, S.Z.; Hu, X.H.; Wang, F.F.; Ye, C.H.U.; Yang, W.Q.; Sheng, X.U.; Song, W.A.N.G.; Wu, L.R.; Yu, H.L.; Miao, H.R.; et al. A stable and major QTL region on chromosome 2 conditions pod shape in cultivated peanut (Arachis hyopgaea L.). J. Integr. Agric. 2023, 22, 2323–2334. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait/Source of Variation | Mean Sum of Squares | ||||
|---|---|---|---|---|---|
| Environment | Replication | Genotypes | G × E | Pooled Error | |
| Degrees of freedom | 1 | 1 | 143 | 143 | 286 |
| Pod yield (kg ha−1) | 9,047,419.28 * | 207,621.32 | 40,307.69 | 61,125.05 *** | 27,562.89 |
| Haulm yield (kg ha−1) | 38,793,685.30 * | 227,604.46 | 56,512.90 * | 55,332.65 | 36,921.52 |
| HPW (g) | 680.68 | 1676.18 | 120.94 | 107.35 *** | 66.61 |
| HSW (g) | 1040.53 | 665.70 | 31.87 | 31.657 *** | 17.71 |
| SP | 7696.37 | 2031.01 | 53.69 | 66.38 *** | 39.92 |
| SCMR | 1948.92 | 439.82 | 46.98 | 44.46 *** | 24.82 |
| Oil content (%) | 470.49 | 1130.28 | 31.43 *** | 12.79 | 9.51 |
| Protein content (%) | 2.08 | 194.32 | 5.22 *** | 2.68 | 1.48 |
| Linoleic acid (%) | 23.66 | 77.20 | 99.13 *** | 26.02 | 17.11 |
| Oleic acid (%) | 526.32 | 867.12 | 137.77 *** | 30.62 | 18.65 |
| Traits | Environment | Parental Means | CD (5%) | Recombinant Inbred Lines | |||||
|---|---|---|---|---|---|---|---|---|---|
| JUG-03 | Valencia-C | RIL | Range | PCV | GCV | H2 | |||
| Mean | |||||||||
| Pod yield | E1 | 580.00 | 890.00 | 266.72 | 675.24 | 580–960 | 25.73 | 16.20 | 39.67 |
| E2 | 299.16 | 690.75 | 371.78 | 420.84 | 121.75–725.16 | 51.37 | 25.29 | 24.25 | |
| Haulm yield | E1 | 465.07 | 297.90 | 171.44 | 434.71 | 229.67–608.5 | 24.98 | 15.03 | 36.20 |
| E2 | 991.50 | 654.5 | 509.05 | 953.75 | 646.5–1418 | 9.22 | 28.53 | 10.45 | |
| HPW | E1 | 66.61 | 94.62 | 22.09 | 78.09 | 61.62–102.06 | 16.37 | 7.95 | 23.57 |
| E2 | 74.50 | 82.00 | 6.14 | 75.93 | 73–86 | 5.50 | 3.67 | 44.56 | |
| HSW | E1 | 30.00 | 39.50 | 11.69 | 34.48 | 28–44 | 20.23 | 10.74 | 28.15 |
| E2 | 30.05 | 32.60 | 1.33 | 31.79 | 29.9–33.2 | 2.78 | 1.79 | 41.58 | |
| SP | E1 | 47.00 | 58.49 | 14.63 | 53.07 | 45.5–63.11 | 15.28 | 6.23 | 16.61 |
| E2 | 52.11 | 66.85 | 9.79 | 60.41 | 50.87–71.5 | 9.54 | 4.86 | 26.03 | |
| SCMR | E1 | 42.50 | 55.35 | 11.44 | 45.32 | 37.5–59.2 | 15.39 | 8.59 | 31.13 |
| E2 | 37.60 | 46.15 | 7.94 | 41.64 | 34.9–48.35 | 11.23 | 5.75 | 26.24 | |
| Oil content | E1 | 46.23 | 42.47 | 7.62 | 44.26 | 38.41–48.94 | 10.11 | 5.14 | 25.87 |
| E2 | 49.28 | 40.41 | 5.73 | 46.08 | 39.21–55.33 | 8.04 | 5.00 | 38.66 | |
| Protein content | E1 | 30.50 | 25.98 | 2.52 | 28.91 | 23.03–31 | 5.04 | 2.43 | 23.21 |
| E2 | 30.22 | 25.75 | 2.27 | 28.78 | 23.03–30.22 | 5.08 | 3.13 | 38.08 | |
| Linoleic acid | E1 | 24.47 | 33.10 | 9.46 | 27.91 | 17.01–36.23 | 20.35 | 10.96 | 29.02 |
| E2 | 22.55 | 30.73 | 9.94 | 27.84 | 12.05–34.59 | 24.74 | 16.9 | 46.65 | |
| Oleic acid | E1 | 42.21 | 52.92 | 11.98 | 49.15 | 38.51–60.86 | 14.3 | 7.24 | 25.65 |
| E2 | 44.14 | 50.32 | 12.9 | 47.24 | 35.59–60.15 | 17.39 | 10.56 | 36.92 | |
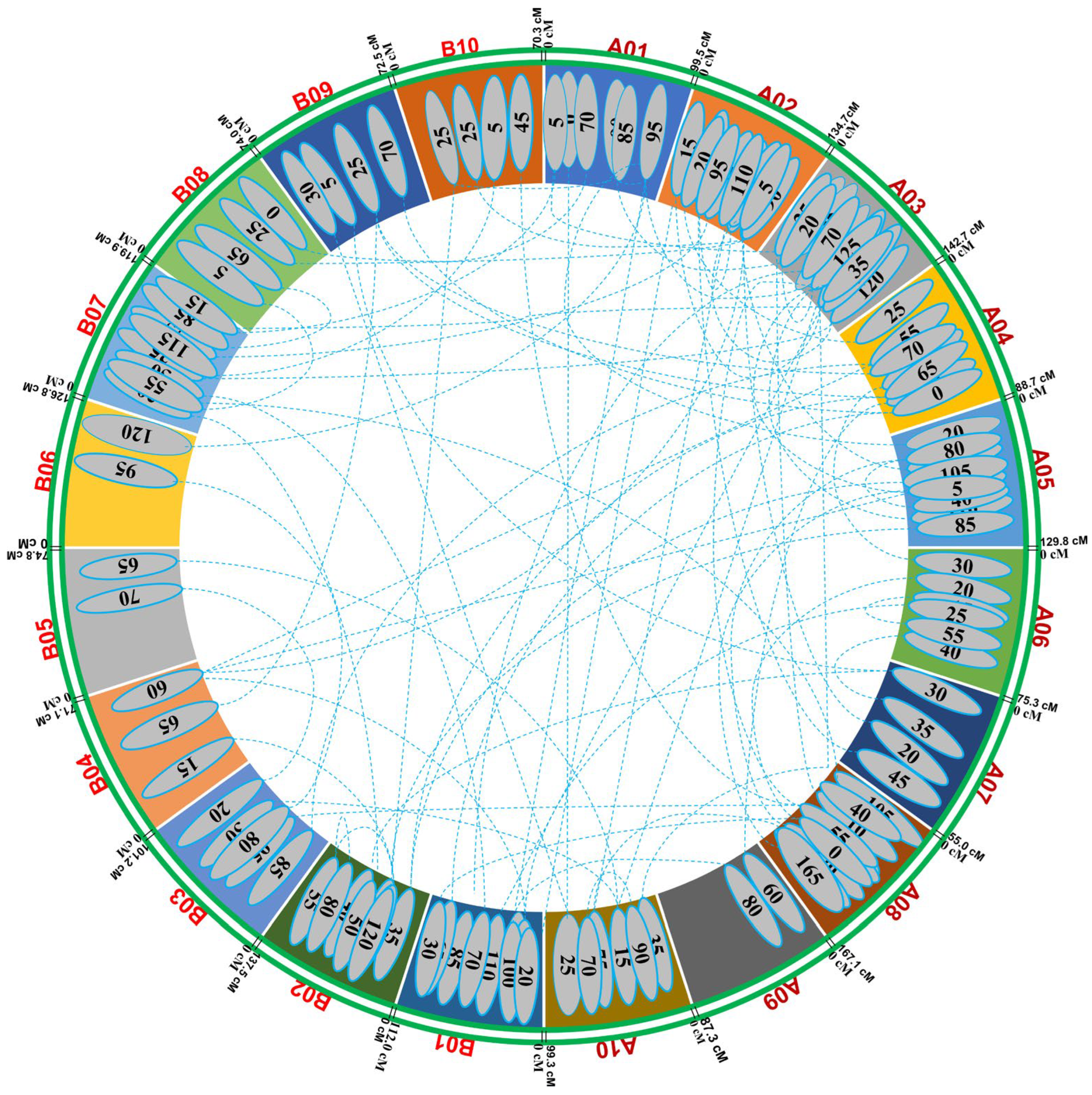
| Chromosome | Mapped Loci | Map Distance (cM) | Inter-Marker Distance (cM) | Map Density (loci/cM) |
|---|---|---|---|---|
| A01 | 58 | 99.50 | 1.72 | 0.58 |
| A02 | 42 | 134.70 | 3.21 | 0.31 |
| A03 | 82 | 142.69 | 1.74 | 0.57 |
| A04 | 61 | 88.70 | 1.45 | 0.69 |
| A05 | 67 | 129.80 | 1.94 | 0.52 |
| A06 | 42 | 75.30 | 1.79 | 0.56 |
| A07 | 22 | 55.00 | 2.50 | 0.40 |
| A08 | 45 | 167.12 | 3.71 | 0.27 |
| A09 | 47 | 87.30 | 1.86 | 0.54 |
| A10 | 92 | 99.36 | 1.08 | 0.93 |
| B01 | 51 | 112.02 | 2.20 | 0.46 |
| B02 | 99 | 137.54 | 1.39 | 0.72 |
| B03 | 89 | 101.21 | 1.14 | 0.88 |
| B04 | 61 | 71.10 | 1.17 | 0.86 |
| B05 | 124 | 74.85 | 0.60 | 1.66 |
| B06 | 91 | 126.80 | 1.39 | 0.72 |
| B07 | 98 | 119.96 | 1.22 | 0.82 |
| B08 | 21 | 74.00 | 3.52 | 0.28 |
| B09 | 20 | 72.50 | 3.63 | 0.28 |
| B10 | 111 | 70.28 | 0.63 | 1.58 |
| Grand Total | 1323 | 2003.13 | 1.89 |
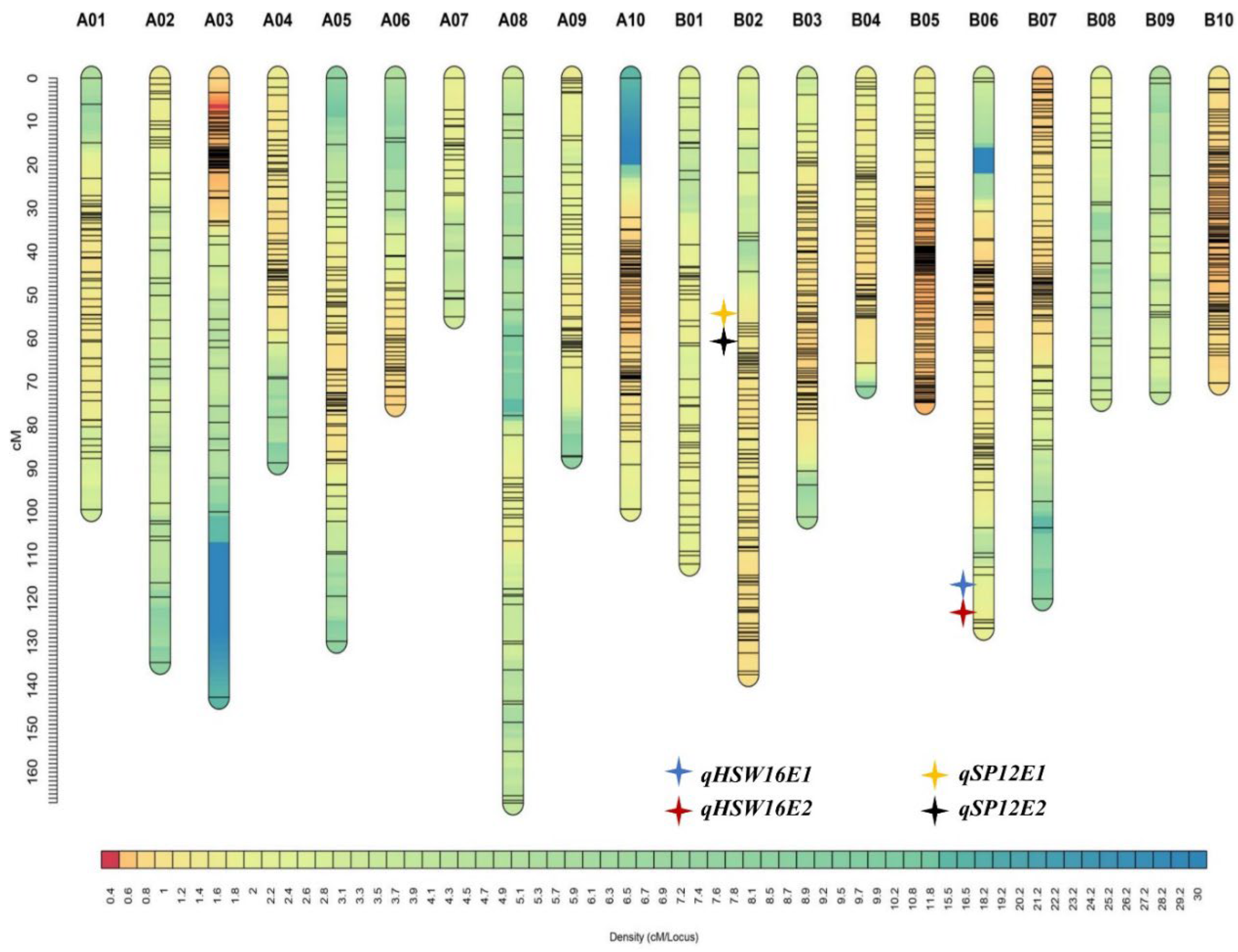
| QTLs | Traits | Env. | Chr. | Pos. (cM) | Marker Interval | LOD | PVE (%) | Additive Effect |
|---|---|---|---|---|---|---|---|---|
| qPODYLD18E1 | Pod yield | E1 | B08 | 28.17 | S18_51822496-S18_134813874 | 4.88 | 6.87 | −155.85 |
| qPODYLD12.2E2 | Pod yield | E2 | B02 | 99.71 | S12_93246314-S12_100066236 | 4.6 | 6.27 | −65.62 |
| qPODYLD12.3E2 | Pod yield | E2 | B02 | 106.01 | S12_110782372-S12_112245066 | 4.78 | 6.53 | −65.04 |
| qHAULMYLD18E1 | Haulm yield | E1 | B08 | 28.61 | S18_51822496-S18_134813874 | 4.34 | 6.09 | −64.16 |
| qHAULMYLD6E1 | Haulm yield | E1 | A06 | 29.16 | S6_1562649-S6_1630272 | 3.64 | 9.55 | 149.88 |
| qHSW12E1 | HSW | E1 | B02 | 57.21 | S12_42838843-S12_73270208 | 4.06 | 5.89 | 9.39 |
| qHSW16E1 | HSW | E1 | B06 | 120.57 | S16_2332048-S16_8231918 | 6.65 | 14.65 | −5.38 |
| qHSW16E2 | HSW | E2 | B06 | 120.55 | S16_2332048-S16_8231918 | 6.11 | 13.87 | −3.31 |
| qSP12E1 | SP | E1 | B02 | 57.23 | S12_42838843-S12_73270208 | 4.36 | 10.98 | 13.95 |
| qSP12E2 | SP | E2 | B02 | 57.21 | S12_42838843-S12_73270208 | 4.43 | 11.65 | 5.44 |
| qSCMR17E2 | SCMR | E2 | B07 | 22.21 | S17_32203546-S17_66864943 | 4.27 | 5.9 | −6.28 |
| qOIL12E1 | Oil content | E1 | B02 | 57.21 | S12_42838843-S12_73270208 | 3.05 | 4.49 | 10.65 |
| qOIL12E2 | Oil content | E2 | B02 | 57.23 | S12_42838843-S12_73270208 | 3.02 | 4.46 | 10.67 |
| qPROTEIN12E2 | Protein content | E2 | B02 | 57.21 | S12_42838843-S12_73270208 | 3.15 | 4.66 | 6.79 |
| qLINOLEIC12E2 | Linoleic acid | E2 | B02 | 64.11 | S12_36421620-S12_118023921 | 3.43 | 5.15 | −3.41 |
| qOLEIC17E1 | Oleic acid | E1 | B07 | 22.21 | S17_32203546-S17_66864943 | 5.76 | 8.09 | −9.09 |
| qOLEIC17E2 | Oleic acid | E2 | B07 | 22.21 | S17_32203546-S17_66864943 | 6.14 | 8.5 | −9.16 |
| Traits | No. of QTLs | LOD Range | PVE % |
|---|---|---|---|
| Pod yield | 9 | 3.08–4.18 | 24.96–38.48 |
| Haulm yield | 15 | 3.00–5.97 | 11.86–54.84 |
| HSW | 4 | 3.04–4.17 | 9.31–20.48 |
| SP | 14 | 3.00–5.72 | 11.72–72.61 |
| SCMR | 8 | 3.00–4.10 | 11.85–22.85 |
| Oil content | 7 | 3.04–4.77 | 11.63–28.23 |
| Protein content | 11 | 8.54–19.99 | 48.31–61.59 |
| Linoleic acid | 2 | 3.02–3.30 | 18.75–26.73 |
| Oleic acid | 7 | 3.08–4.38 | 13.07–50.08 |
| Traits | QTL Name | Gene Location | Gene Model | Nearest SNP (bp) | Functional Annotation |
|---|---|---|---|---|---|
| SP | qSP12E1 and qSP12E2 | Araip.B02 | Araip.6MG4Z | 67,500,914 | Disease resistance protein |
| Araip.DH675 | 42,817,549 | Serine/threonine-protein phosphatase | |||
| Araip.5HJ7H | 67,500,914 | Protein MIZU-KUSSEI 1 | |||
| Araip.7QC0C | 42,817,549 | Actin-related protein | |||
| HSW | qHSW16E1 and qHSW16E2 | Araip.B06 | Araip.49T7Y | 8,393,784 | Protein kinase superfamily protein |
| Araip.5E3CZ | 1,562,649 | Transcription factor bHLH68 | |||
| Araip.CXF88 | 271,015 | CBS domain-containing protein | |||
| Araip.GWR7V | 2,818,988 | Seed maturation protein | |||
| Araip.LE5CL | 1,562,649 | Ethylene-responsive transcription factor | |||
| Araip.UY42T | 2,697,960 | Isopentenyltransferase 3 | |||
| Araip.WM0UU | 7,821,084 | Cytochrome P450 superfamily protein | |||
| Araip.60J6J | 2,818,988 | GTP binding elongation factor | |||
| Araip.CY9QC | 8,393,784 | Ribosomal protein L19e family protein | |||
| Araip.SKT5W | 1,562,649 | NAD+ ADP-ribosyltransferase |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joshi, P.; Soni, P.; Sharma, V.; Manohar, S.S.; Kumar, S.; Sharma, S.; Pasupuleti, J.; Vadez, V.; Varshney, R.K.; Pandey, M.K.; et al. Genome-Wide Mapping of Quantitative Trait Loci for Yield-Attributing Traits of Peanut. Genes 2024, 15, 140. https://doi.org/10.3390/genes15020140
Joshi P, Soni P, Sharma V, Manohar SS, Kumar S, Sharma S, Pasupuleti J, Vadez V, Varshney RK, Pandey MK, et al. Genome-Wide Mapping of Quantitative Trait Loci for Yield-Attributing Traits of Peanut. Genes. 2024; 15(2):140. https://doi.org/10.3390/genes15020140
Chicago/Turabian StyleJoshi, Pushpesh, Pooja Soni, Vinay Sharma, Surendra S. Manohar, Sampath Kumar, Shailendra Sharma, Janila Pasupuleti, Vincent Vadez, Rajeev K. Varshney, Manish K. Pandey, and et al. 2024. "Genome-Wide Mapping of Quantitative Trait Loci for Yield-Attributing Traits of Peanut" Genes 15, no. 2: 140. https://doi.org/10.3390/genes15020140
APA StyleJoshi, P., Soni, P., Sharma, V., Manohar, S. S., Kumar, S., Sharma, S., Pasupuleti, J., Vadez, V., Varshney, R. K., Pandey, M. K., & Puppala, N. (2024). Genome-Wide Mapping of Quantitative Trait Loci for Yield-Attributing Traits of Peanut. Genes, 15(2), 140. https://doi.org/10.3390/genes15020140