2. Methods
We generalize the MWC model using the framework of Zhao and Satten [
6] by adding an interaction bias
to the log-linear model:
where
is the true relative abundance of taxon
j in sample
i,
is the expected value of the observed relative abundance,
is the main effect bias for taxon
j in the MWC model, and
is the sample-specific normalization factor that ensures the compositional constraint on
. We followed [
6] to introduce the effects of covariates
on taxon
j by replacing
in (
1) with
, where
contains the effect sizes, and
is the true relative abundance of taxon
j when
. Note that, similar to all compositional analysis methods, the effect size parameters
here characterize differences in “absolute” abundances rather than “relative” abundances; relative abundances are further modulated by the normalization factor
which is a function of all the
s. The interaction
determines the extent to which taxon
affects the bias factor for taxon
j. The assumption underlying the MWC model is
for all
j and
. We adopt the following model for generating the interaction biases relative to the main effect biases in our simulation studies:
where
is a constant for all taxa pairs that we call the magnitude of interaction biases, and
is a non-negative error term with a mean of one.
The value of
and the minus sign in (
2) reflect the findings of Zhao and Satten [
6], who considered a model similar to (
1) but with an interaction that depended on the presence of a taxon rather than its relative abundance. In particular, they considered the model
where
determines the extent to which the
presence of taxon
affects the bias factor for taxon
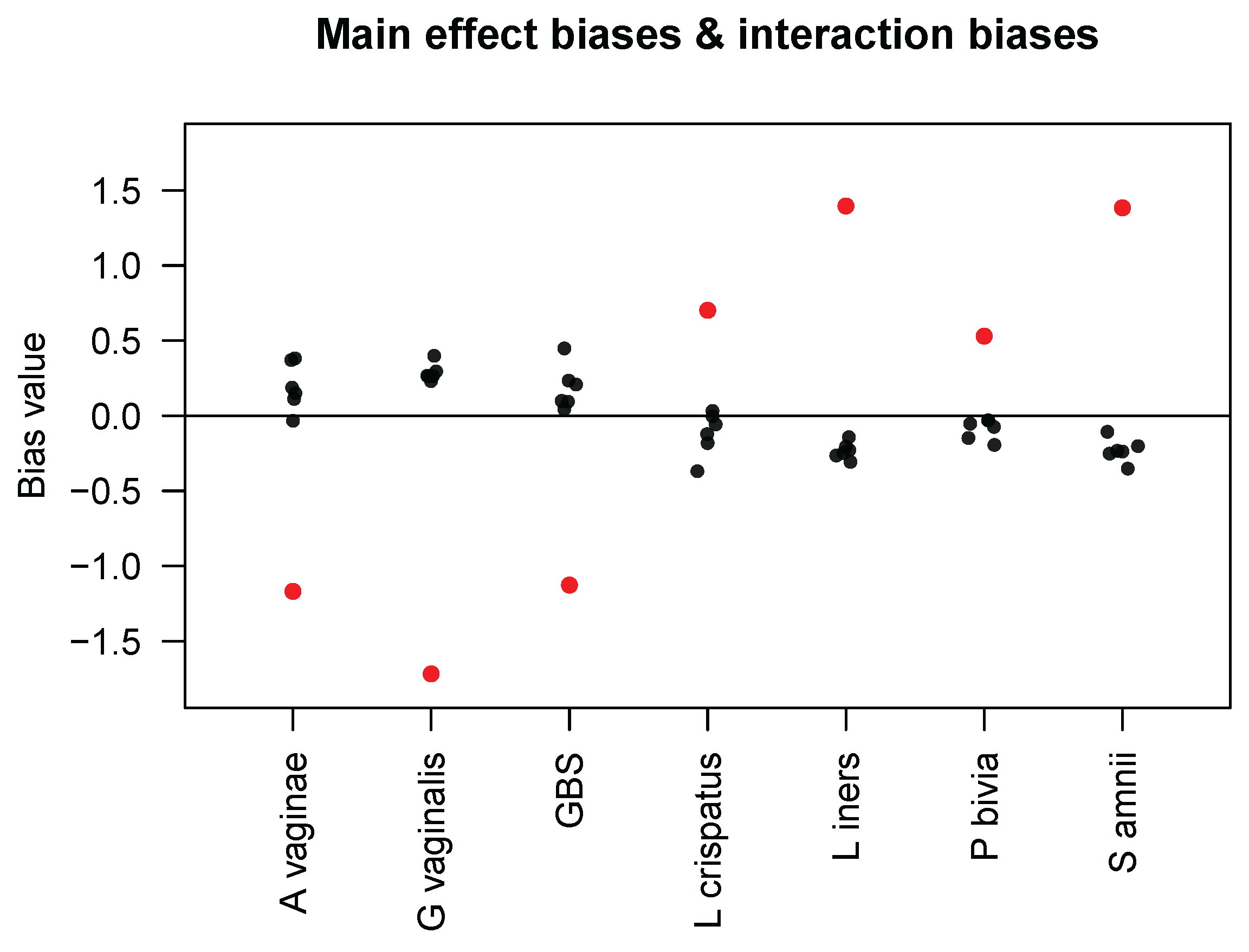
j. Using data from all seven taxa in the Brooks mock community samples, they estimated the main effect biases
s and interaction biases
s and presented the results in their Table 4. For the convenience of our readers, we display their results in
Figure 1. First, we observe that the interaction biases have smaller magnitudes than the main effect biases; we calculated the mean of the interaction–main effect ratios (i.e.,
) to be 0.204. Because
in model (
3) corresponds to the product
in our model (
1) and because most of the Brooks samples have three taxa with equal proportions, i.e.,
, we obtained the (approximate) mean of
and, thus, an estimate for
of 0.612. In our simulations, we varied the value of
from 0 to 4, which includes our empirical estimate of 0.612 while also extending the range of
to allow for stronger interaction biases than those found in the Brooks data. In addition,
Figure 1 shows that the interaction bias in taxon
j caused by taxon
has an opposite sign to the sign of the main effect bias in taxon
, except for some very small interaction biases. This implies that if taxon
has a measured abundance higher than its true abundance, it is associated with decreased measured abundances of other taxa, which seems plausible if we assume that only a relatively constant total number of amplicons are sequenced. Finally, interaction biases are not necessarily symmetric between a pair of taxa, as also noted by Zhao and Satten [
6], so our model (
2) also does not guarantee such symmetry.
Our simulation studies were based on models (
1) and (
2) and data on 856 taxa of the upper-respiratory tract (URT) microbiome by Charlson et al. [
15]. We considered three scenarios for the distribution of interaction biases. In the first scenario (referred to as S-nondiff), we sampled
from
for all taxa pairs
j and
. In the second scenario (referred to as S-diff-causal), we modified S-nondiff to sample
from
when taxon
j was “causal” (i.e., associated with the trait of interest) and for all
. Both distributions have a mean of 0.5; however,
has one mode at 0.5, whereas
has two modes 0 and 1 and, hence, a variance of larger than
. In the third scenario (referred to as S-diff-half), we used
for half of the randomly selected taxa
js and
for the remaining half of the taxa. Unlike S-nondiff, S-diff-causal has a modest (but trait-related) variation in
, while S-diff-half has a large variation in
that is not trait-related. The distribution of the bias factor due to taxon–taxon interactions,
, in each taxon
j across all samples is displayed in
Figures S1 and S2 and has the following features. In all scenarios and for all taxa
js, the mean of
was approximately zero due to the averaging of contributions with different directionalities. The variance of
increased as
increased at a given
. S-nondiff and S-diff-causal had the same
values at the null taxa and different
values at the causal taxa. Finally, in S-diff-half,
had a larger variance in one half of the taxa than in the other half of the taxa.
Additional details of the simulation settings generally follow the simulations in [
5]. Specifically, we considered both binary and continuous traits of interest without any confounding covariates; we also considered a binary confounder when the trait was binary. We used the two sets of “causal” taxa (i.e., taxa that are associated with the trait) that were used in [
5] and referred to as M1 and M2. In M1, 20 taxa were randomly sampled to be causal from the set of taxa having mean relative abundances in the URT data [
15] greater than 0.005 (but excluding the most prevalent taxon). In M2, the top five most abundant taxa (having mean relative abundances of 0.105, 0.062, 0.054, 0.050, and 0.049) were selected to be causal. For simulations with a binary confounder, we assumed that the confounder was associated with 20 taxa under M1 (10 sampled at random from the causal taxa and 10 from the null taxa) and 5 taxa under M2 (2 from the causal taxa and 3 from the null taxa). We set the sample size to 100. Let
denote the trait and
denote the confounder for the
ith sample so that
. To generate a binary trait, we selected an equal number of samples with
and
. When a binary confounder was present, we drew
from
in samples with
and from
in samples with
. To generate a continuous trait, we sampled
from
. To simulate the read count data for the 856 taxa, we first sampled the baseline relative abundances
of all taxa for each sample from
, in which
and
took the estimated mean and overdispersion (0.02) from fitting the Dirichlet–Multinomial model to the URT data. We formed the
true relative abundances
for all taxa by spiking the causal taxon
with an
fold change, spiking the confounder-associated taxon
with an
fold change, and normalizing the relative abundances, so that
We formed the
observed relative abundances
by additionally multiplying the bias factor
, so that
Note that
for null taxa and
for confounder-independent taxa. For simplicity, we set
, a common effect size of the trait, on all causal taxa. We generated the main effect bias
from
, which gave a range between 0.2 and 5 for most (95%) fold changes (
) caused by the main effect bias. The scheme for generating the interaction bias
has been described earlier. Finally, we generated the taxon count data using the Multinomial model with the mean
and a library size sampled from an
N(10,000, (10,000/3
distribution that was left-truncated at 2000.
Following [
5], we applied the following compositional analysis methods: LOCOM, ANCOM, ANCOM-BC, ALDEx2, DACOMP (v1.26), WRENCH, Wilcox-alr-half, and Wilcox-alr-one. The latter two are Wilcoxon rank-sum tests of additive-log-ratio transformed count data after adding pseudocount 0.5 or 1 to all count data and using the most abundant null taxon (known in simulated data) as the reference; the
p-values were adjusted for multiple testing by the Benjamini–Hochberg [
16] procedure. In addition, we included two newly developed methods, fastANCOM and LinDA. LOCOM requires that the taxa present in fewer than 20% of samples are filtered out. Recall that, since LOCOM fits logistic regression models to taxa count data, zero read counts are naturally handled as possible outcome values. We applied all methods to each replicate of simulated data after using this filter. For ANCOM, ANCOM-BC, fastANCOM, and LinDA, we also applied them to data using their own filter with 10% presence as the cutoff and denoted them as ANCOM
o, ANCOM-BC
o, fastANCOM
o, and LinDA
o. The empirical FDR and sensitivity (proportion of truly causal taxa that were detected) were evaluated at the nominal level of 20% based on 1000 replicates of data. A relatively high nominal FDR level was chosen due to the small numbers of causal taxa in both M1 and M2.
3. Results
The empirical FDR and sensitivity of all aforementioned compositional analysis methods for detecting the “causal” taxa (i.e., taxa that are associated with the trait), for simulations with a binary trait and no confounder under scenarios M1 and M2, are displayed in
Figure 2 and
Figure 3, respectively (results for simulations with confounders and continuous traits showed similar patterns and are therefore deferred to
Figures S3–S6 in Supplementary Materials). Note that
corresponds to no interaction bias at any taxa (i.e., main effect biases only), and
corresponds to no differential abundance at any taxa (i.e., the global null). In all cases, the FDR of LOCOM remained at or close to the nominal level as long as the magnitude of the interaction bias
, which is substantially larger than the empirical value of 0.612 observed in the Brooks data [
6]; under the global null in particular, LOCOM always controlled the FDR regardless of the value of
. It was only when both
and
became unrealistically large that we observed moderate inflation in the FDR of LOCOM. The FDR inflation of LOCOM was similar in S-nondiff and S-diff-causal because the interaction biases were similarly distributed at the majority of taxa, which were null taxa; the inflation was (slightly) larger in S-diff-half because the interaction biases had the largest variability among the three scenarios at a given
. The interaction biases caused some loss of sensitivity for LOCOM, but the drop was relatively small, and LOCOM maintained the highest sensitivity among all methods in all of our simulation scenarios. The results of the other methods showed a similar trend to LOCOM with the FDR increasing and the sensitivity decreasing as
increased, as well as an increase in FDR inflation as
increased.
The other methods had very different performances even when there were only main effect biases (). ANCOM-BC and fastANCOM performed the best among the other methods, controlling the FDR for the full range of values we have considered when was small and having similar FDR inflation as LOCOM when both and became large; however, this performance came at the cost of a substantially lower sensitivity than that of LOCOM. ANCOM had a moderately inflated FDR when was small but performed better when was increased; nevertheless, its sensitivity was among the lowest. ALDEx2 had an inflated FDR and poor sensitivity when was large in M1 but not in M2. WRENCH, Wilcox-alr-half, and Wilcox-alr-one had highly inflated FDRs except in the global null case, and LinDA had a highly inflated FDR in all cases including the global null. These results were based on data after applying the filter of rare taxa as recommended by LOCOM; in general, ANCOM, ANCOM-BC, fastANCOM, and LinDA had worse FDR control when a less stringent filter was applied. Note that, unlike the LOCOM paper containing results from the older version (v1.1) of DACOMP, we used the latest version (v1.26) here, which yielded a highly inflated FDR and low sensitivity in M2 because some causal taxa were incorrectly selected to be among the reference set.



{kind=link}
{kind=link}
{kind=link}