
Five Regions of the Pea Genome Co-Control Partial Resistance to D. pinodes, Tolerance to Frost, and Some Architectural or Phenological Traits

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Fungal and Plant Material
2.2. Partial Resistance to D. pinodes Assessed on Whole Plantlets and Detached Stipules under Controlled Conditions
2.3. Architectural Traits Assessments on Whole Plantlets or Detached Stipules under Controlled Conditions
2.4. Frost Tolerance
2.5. Statistical Analyses for D. pinodes Disease Severity, Architectural Traits, and Frost Tolerance
2.6. SNP KASP™ Genotyping Assays
2.7. Construction of Three High-Resolution Individual RIL Maps
2.8. QTL Analyses
3. Results
3.1. High-Density Individual Genetic Maps
3.2. Phenotyping D. pinodes Severity and Architectural Traits Data on RIL Populations
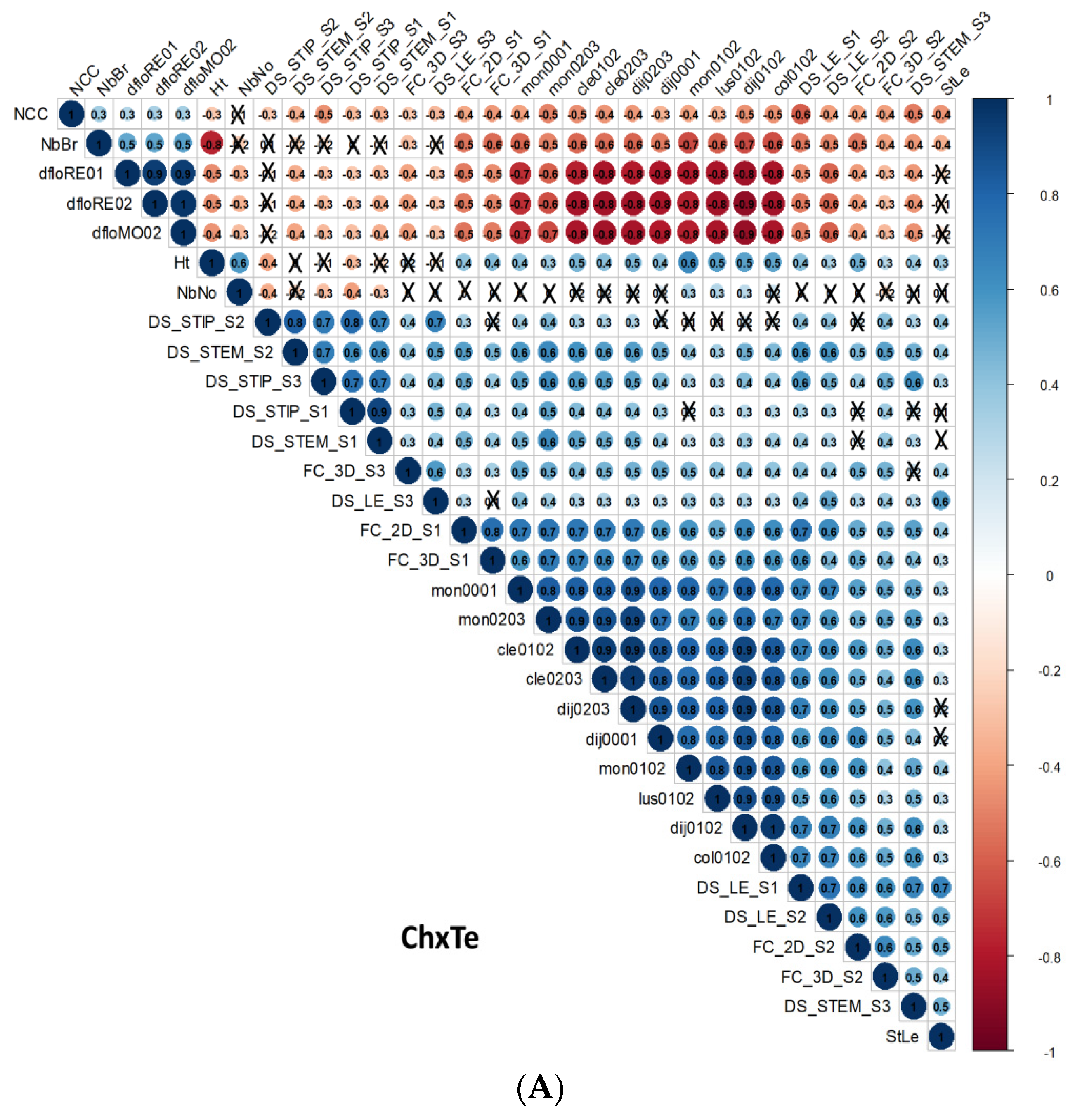
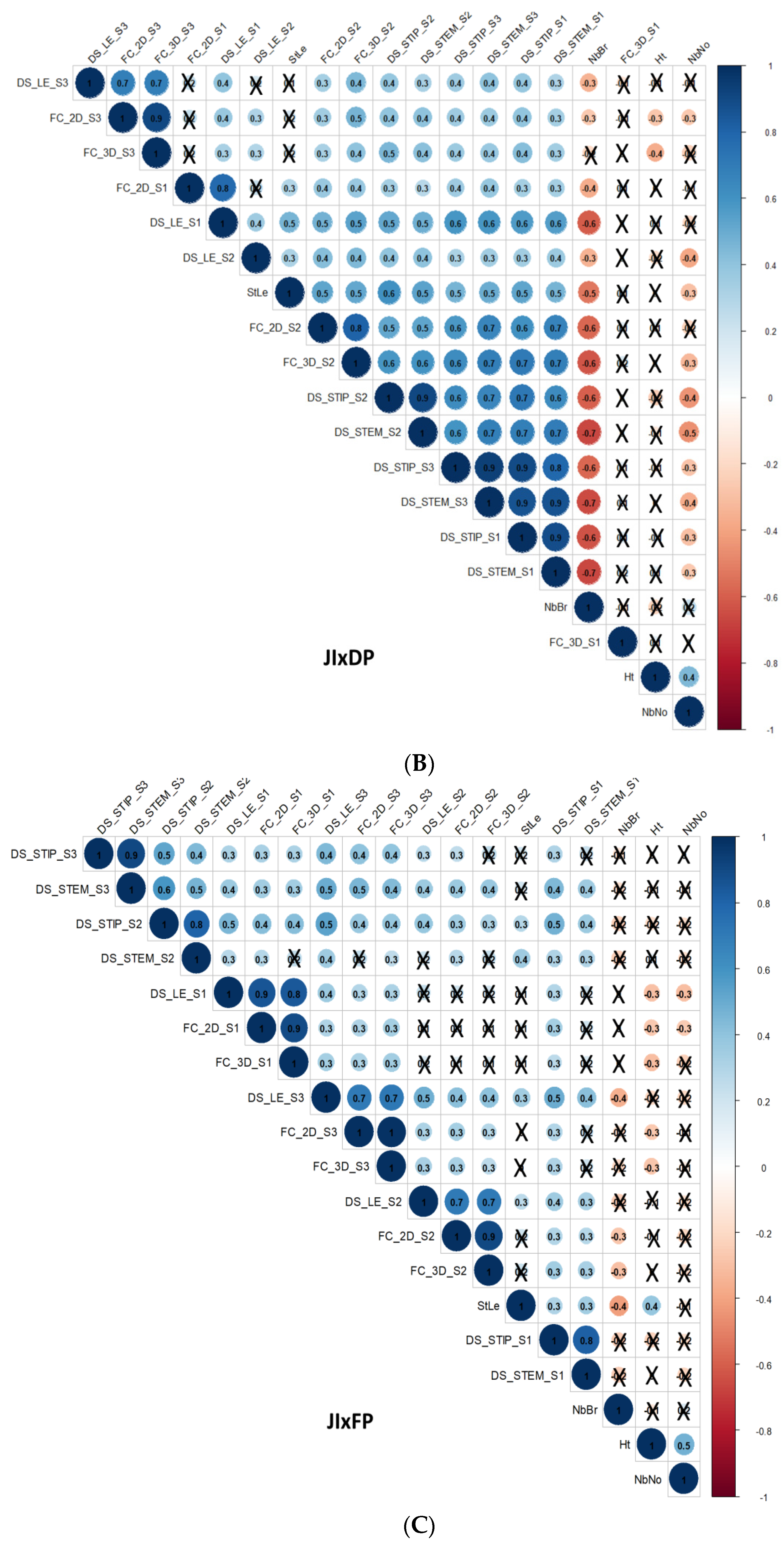
3.3. Correlations
3.3.1. Correlations between D. pinodes Disease Severity Traits
3.3.2. Correlations between Architectural and Phenological Traits
3.3.3. Correlations between D. pinodes Disease Severity Traits and Phenological or Architectural Traits
3.3.4. Correlations between Environments, Assessment Methods for Frost Tolerance, Architectural and Phenological Traits, and Disease Severity Assessments within the ChxTe Population
3.4. QTLs Controlling D. pinodes Disease Severity, Frost Tolerance, and Some Architectural and Phenological Traits
3.4.1. QTL Detection on Each Individual Map
3.4.2. QTL Projection on the Composite Map and metaQTL Analysis by Trait
3.4.3. Joint metaQTL Analysis
4. Discussion
4.1. New HD Genetic Maps and Phenotyping Data Improve QTL Detection Accuracy and Robustness
4.2. Genomic Regions Controlling D. pinodes Disease Severity Are Consistent across Strains and Organs, However, Depend upon Resistance Sources and Mapping Populations
4.3. Co-Control of D. pinodes Disease Severity, Frost Tolerance, and Architectural or Phenological Traits in Five Regions of the Pea Genome
4.3.1. D. pinodes Resistance and Frost Tolerance Were Associated with a Delayed Flowering and a Higher Number of Basal Branches
4.3.2. D. pinodes Resistance and Frost Tolerance Are Associated with Small Stipules
4.3.3. The Major Gene Le Colocalizes with Both D. pinodes Resistance and Frost Tolerance QTLs
4.3.4. The Major Gene Hr Colocalizes with Both D. pinodes Resistance and Frost Tolerance QTLs
4.3.5. Underlying Positional, Expressional, or Functional Candidate Genes in MDAFs’ Confidence Intervals
4.4. Impact of Colocalizations for Breeding
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tivoli, B.; Calonnec, A.; Richard, B.; Ney, B.; Andrivon, D. Current knowledge on plant/canopy architectural traits that reduce the expression and development of epidemics. Eur. J. Plant Pathol. 2013, 135, 471–478. [Google Scholar] [CrossRef]
- Ney, B.; Bancal, M.-O.; Bancal, P.; Bingham, I.; Foulkes, J.; Gouache, D.; Paveley, N.; Smith, J. Crop architecture and crop tolerance to fungal diseases and insect herbivory. Mechanisms to limit crop losses. Eur. J. Plant Pathol. 2013, 135, 561–580. [Google Scholar] [CrossRef]
- Richard, B.; Jumel, S.; Rouault, F.; Tivoli, B. Influence of plant stage and organ age on the receptivity of Pisum sativum to Mycosphaerella pinodes. Eur. J. Plant Pathol. 2012, 132, 367–379. [Google Scholar] [CrossRef]
- Larkin, R.P.; Fravel, D.R. Efficacy of various fungal and bacterial biocontrol organisms for control of Fusarium wilt of tomato. Plant Dis. 1998, 82, 1022–1028. [Google Scholar] [CrossRef] [PubMed]
- Simonetta, S.; Avidano, L.; Berta, G. Morphogenetic effects induced by pathogenic and non pathogenic Rhizoctonia solani Kühn strains on tomato roots. Caryologia 2007, 60, 141–145. [Google Scholar] [CrossRef]
- Kraft, J.M.; Boge, W. Root characteristics in pea in relation to compaction and Fusarium root rot. Plant Dis. 2001, 85, 936–940. [Google Scholar] [CrossRef]
- Bonhomme, M.; André, O.; Badis, Y.; Ronfort, J.; Burgarella, C.; Chantret, N.; Prosperi, J.M.; Briskine, R.; Mudge, J.; Debellé, F. High-density genome-wide association mapping implicates an F-box encoding gene in M edicago truncatula resistance to A phanomyces euteiches. New Phytol. 2014, 201, 1328–1342. [Google Scholar] [CrossRef]
- Omran, A.O.; Atkins, I.M.; Gilmore, E.C., Jr. Heritability of Cold Hardness in Flax (Linum usitatissinum L.). Crop Sci. 1968, 8, 716–719. [Google Scholar] [CrossRef]
- Rikin, A.; Waldman, M.; Richmond, A.E.; Dovrat, A. Hormonal Regulation of Morphogenesis and Cold-resistance: I. Modifications by abscisic acid and by gibberellic acid in alfalfa (Medicago sativa L.) Seedlings. J. Exp. Bot. 1975, 26, 175–183. [Google Scholar] [CrossRef]
- Murray, G.; Swensen, J. Winterhardiness of pea. Pisum Genet. 1991, 23, 4–13. [Google Scholar]
- Karlova, R.; Boer, D.; Hayes, S.; Testerink, C. Root plasticity under abiotic stress. Plant Physiol. 2021, 187, 1057–1070. [Google Scholar] [CrossRef] [PubMed]
- Hyles, J.; Bloomfield, M.T.; Hunt, J.R.; Trethowan, R.M.; Trevaskis, B. Phenology and related traits for wheat adaptation. Heredity 2020, 125, 417–430. [Google Scholar] [CrossRef]
- Bhat, K.A.; Mahajan, R.; Pakhtoon, M.M.; Urwat, U.; Bashir, Z.; Shah, A.A.; Agrawal, A.; Bhat, B.; Sofi, P.A.; Masi, A.; et al. Low Temperature Stress Tolerance: An Insight into the Omics Approaches for Legume Crops. Front. Plant Sci. 2022, 13, 888710. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, N.J.; Urwin, P.E. The interaction of plant biotic and abiotic stresses: From genes to the field. J. Exp. Bot. 2012, 63, 3523–3543. [Google Scholar] [CrossRef] [PubMed]
- Saijo, Y.; Loo, E.P.-I. Plant immunity in signal integration between biotic and abiotic stress responses. New Phytol. 2020, 225, 87–104. [Google Scholar] [CrossRef]
- Bhogireddy, S.; Mangrauthia, S.K.; Kumar, R.; Pandey, A.K.; Singh, S.; Jain, A.; Budak, H.; Varshney, R.K.; Kudapa, H. Regulatory non-coding RNAs: A new frontier in regulation of plant biology. Funct. Integr. Genom. 2021, 21, 313–330. [Google Scholar] [CrossRef]
- Zhang, H.; Sonnewald, U. Differences and commonalities of plant responses to single and combined stresses. Plant J. 2017, 90, 839–855. [Google Scholar] [CrossRef]
- Wisser, R.J.; Balint-Kurti, P.J.; Nelson, R.J. The Genetic Architecture of Disease Resistance in Maize: A Synthesis of Published Studies. Phytopathology 2006, 96, 120–129. [Google Scholar] [CrossRef]
- Barmukh, R.; Roorkiwal, M.; Dixit, G.P.; Bajaj, P.; Kholova, J.; Smith, M.R.; Chitikineni, A.; Bharadwaj, C.; Sheshshayee, M.S.; Rathore, A.; et al. Characterization of “QTL-hotspot” introgression lines reveals physiological mechanisms and candidate genes associated with drought adaptation in chickpea. J. Exp. Bot. 2022, 73, 7255–7272. [Google Scholar] [CrossRef]
- Poland, J.A.; Balint-Kurti, P.J.; Wisser, R.J.; Pratt, R.C.; Nelson, R.J. Shades of gray: The world of quantitative disease resistance. Trends Plant Sci. 2009, 14, 21–29. [Google Scholar] [CrossRef]
- Le May, C.; Schoeny, A.; Tivoli, B.; Ney, B. Improvement and validation of a pea crop growth model to simulate the growth of cultivars infected with Ascochyta blight (Mycosphaerella pinodes). Eur. J. Plant Pathol. 2005, 112, 1–12. [Google Scholar] [CrossRef]
- Bénézit, M.; Biarnès, V.; Jeuffroy, M.-H. Impact of climate and diseases on pea yields: What perspectives with climate change? OCL 2017, 24, D103. [Google Scholar] [CrossRef]
- Timmerman-Vaughan, G.M.; Frew, T.J.; Russell, A.C.; Khan, T.; Butler, R.; Gilpin, M.; Murray, S.; Falloon, K. QTL Mapping of Partial Resistance to Field Epidemics of Ascochyta Blight of Pea. Crop Sci. 2002, 42, 2100–2111. [Google Scholar] [CrossRef]
- Prioul, S.; Frankewitz, A.; Deniot, G.; Morin, G.; Baranger, A. Mapping of quantitative trait loci for partial resistance to Mycosphaerella pinodes in pea (Pisum sativum L.), at the seedling and adult plant stages. Ther. Appl. Genet. 2004, 108, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Timmerman-Vaughan, G.M.; Mills, A.; Whitfield, C.; Frew, T.; Butler, R.; Murray, S.; Lakeman, M.; McCallum, J.; Russell, A.; Wilson, D. Linkage mapping of QTL for seed yield, yield components, and developmental traits in pea. Crop Sci. 2005, 45, 1336–1344. [Google Scholar] [CrossRef]
- Jha, A.B.; Tar’an, B.; Stonehouse, R.; Warkentin, T.D. Identification of QTLs Associated with Improved Resistance to Ascochyta Blight in an Interspecific Pea Recombinant Inbred Line Population. Crop Sci. 2016, 56, 2926–2939. [Google Scholar] [CrossRef]
- Fondevilla, S.; Almeida, N.F.; Satovic, Z.; Rubiales, D.; Vaz Patto, M.C.; Cubero, J.I.; Torres, A.M. Identification of common genomic regions controlling resistance to Mycosphaerella pinodes, earliness and architectural traits in different pea genetic backgrounds. Euphytica 2011, 182, 43–52. [Google Scholar] [CrossRef]
- Carrillo, E.; Satovic, Z.; Aubert, G.; Boucherot, K.; Rubiales, D.; Fondevilla, S. Identification of quantitative trait loci and candidate genes for specific cellular resistance responses against Didymella pinodes in pea. Plant Cell Rep. 2014, 33, 1133–1145. [Google Scholar] [CrossRef]
- Klein, A.; Houtin, H.; Rond, C.; Marget, P.; Jacquin, F.; Boucherot, K.; Huart, M.; Riviere, N.; Boutet, G.; Lejeune-Henaut, I.; et al. QTL analysis of frost damage in pea suggests different mechanisms involved in frost tolerance. Ther. Appl. Genet. 2014, 127, 1319–1330. [Google Scholar] [CrossRef]
- Lejeune-Hénaut, I.; Hanocq, E.; Béthencourt, L.; Fontaine, V.; Delbreil, B.; Morin, J.; Petit, A.; Devaux, R.; Boilleau, M.; Stempniak, J.J.; et al. The flowering locus Hr colocalizes with a major QTL affecting winter frost tolerance in Pisum sativum L. Ther. Appl. Genet. 2008, 116, 1105–1116. [Google Scholar] [CrossRef]
- Dumont, E.; Fontaine, V.; Vuylsteker, C.; Sellier, H.; Bodèle, S.; Voedts, N.; Devaux, R.; Frise, M.; Avia, K.; Hilbert, J.-L.; et al. Association of sugar content QTL and PQL with physiological traits relevant to frost damage resistance in pea under field and controlled conditions. Ther. Appl. Genet. 2009, 118, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Onfroy, C.; Baranger, A.; Tivoli, B. Biotic factors affecting the expression of partial resistance in pea to ascochyta blight in a detached stipule assay. In Ascochyta Blights of Grain Legumes; Springer: Berlin/Heidelberg, Germany, 2007; pp. 13–27. [Google Scholar]
- Prioul, S.; Onfroy, C.; Tivoli, B.; Baranger, A. Controlled environment assessment of partial resistance to Mycosphaerella pinodes in pea (Pisum sativum L.) seedlings. Euphytica 2003, 131, 121–130. [Google Scholar] [CrossRef]
- Onfroy, C.; Tivoli, B.; Corbiere, R.; Bouznad, Z. Cultural, molecular and pathogenic variability of Mycosphaerella pinodes and Phoma medicaginis var. pinodella isolates from dried pea (Pisum sativum) in France. Plant Pathol. 1999, 48, 218–229. [Google Scholar] [CrossRef]
- Shaner, G.; Finney, R. The effect of nitrogen fertilization on the expression of slow-mildewing resistance in Knox wheat. Phytopathology 1977, 67, 1051–1056. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2013. [Google Scholar]
- Boutet, G.; Alves Carvalho, S.; Falque, M.; Peterlongo, P.; Lhuillier, E.; Bouchez, O.; Lavaud, C.; Pilet-Nayel, M.-L.; Rivière, N.; Baranger, A. SNP discovery and genetic mapping using genotyping by sequencing of whole genome genomic DNA from a pea RIL population. BMC Genom. 2016, 17, 121. [Google Scholar] [CrossRef]
- Tayeh, N.; Aluome, C.; Falque, M.; Jacquin, F.; Klein, A.; Chauveau, A.; Bérard, A.; Houtin, H.; Rond, C.; Kreplak, J.; et al. Development of two major resources for pea genomics: The GenoPea 13.2K SNP Array and a high-density, high-resolution consensus genetic map. Plant J. 2015, 84, 1257–1273. [Google Scholar] [CrossRef]
- Duarte, J.; Riviere, N.; Baranger, A.; Aubert, G.; Burstin, J.; Cornet, L.; Lavaud, C.; Lejeune-Henaut, I.; Martinant, J.-P.; Pichon, J.-P.; et al. Transcriptome sequencing for high throughput SNP development and genetic mapping in Pea. BMC Genom. 2014, 15, 126. [Google Scholar] [CrossRef]
- Alves-Carvalho, S.; Aubert, G.; Carrère, S.; Cruaud, C.; Brochot, A.-L.; Jacquin, F.; Klein, A.; Martin, C.; Boucherot, K.; Kreplak, J.; et al. Full-length de novo assembly of RNA-seq data in pea (Pisum sativum L.) provides a gene expression atlas and gives insights into root nodulation in this species. Plant J. 2015, 84, 1–19. [Google Scholar] [CrossRef]
- Kreplak, J.; Madoui, M.-A.; Cápal, P.; Novák, P.; Labadie, K.; Aubert, G.; Bayer, P.E.; Gali, K.K.; Syme, R.A.; Main, D.; et al. A reference genome for pea provides insight into legume genome evolution. Nat. Genet. 2019, 51, 1411–1422. [Google Scholar] [CrossRef]
- Semagn, K.; Babu, R.; Hearne, S.; Olsen, M. Single nucleotide polymorphism genotyping using Kompetitive Allele Specific PCR (KASP): Overview of the technology and its application in crop improvement. Mol. Breed. 2014, 33, 1–14. [Google Scholar] [CrossRef]
- Loridon, K.; McPhee, K.; Morin, J.; Dubreuil, P.; Pilet-Nayel, M.L.; Aubert, G.; Rameau, C.; Baranger, A.; Coyne, C.; Lejeune-Hènaut, I.; et al. Microsatellite marker polymorphism and mapping in pea (Pisum sativum L.). Ther. Appl. Genet. 2005, 111, 1022–1031. [Google Scholar] [CrossRef]
- Bordat, A.; Savois, V.; Nicolas, M.; Salse, J.; Chauveau, A.; Bourgeois, M.; Potier, J.; Houtin, H.; Rond, C.; Murat, F.; et al. Translational Genomics in Legumes Allowed Placing In Silico 5460 Unigenes on the Pea Functional Map and Identified Candidate Genes in Pisum sativum L. G3 Genes Genomes Genet. 2011, 1, 93–103. [Google Scholar] [CrossRef] [PubMed]
- De Givry, S.; Bouchez, M.; Chabrier, P.; Milan, D.; Schiex, T. CARHTA GENE: Multipopulation integrated genetic and radiation hybrid mapping. Bioinformatics 2005, 21, 1703–1704. [Google Scholar] [CrossRef] [PubMed]
- Haldane, J. The combination of linkage values, and the calculation of distance between the loci of linked factors. J. Genet. 1919, 8, 299–309. [Google Scholar]
- Voorrips, R.E. MapChart: Software for the Graphical Presentation of Linkage Maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef]
- Broman, K.W.; Wu, H.; Arends, D.; Corty, R.; Flutre, T.; Jansen, R.; Prins, P.; Ronnegard, L.; Shah, R.; Shannon, L. Package ‘qtl’. 2019. Available online: https://rqtl.org (accessed on 30 June 2023).
- Lander, E.S.; Botstein, D. Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 1989, 121, 185–199. [Google Scholar] [CrossRef]
- Sosnowski, O.; Charcosset, A.; Joets, J. BioMercator V3, an upgrade of genetic map compilation and quantitative trait loci meta-analysis algorithms. Bioinformatics 2012, 28, 2082–2083. [Google Scholar] [CrossRef] [PubMed]
- Goffinet, B.; Gerber, S. Quantitative Trait Loci: A Meta-analysis. Genetics 2000, 155, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Beji, S.; Fontaine, V.; Devaux, R.; Thomas, M.; Negro, S.S.; Bahrman, N.; Siol, M.; Aubert, G.; Burstin, J.; Hilbert, J.-L.; et al. Genome-wide association study identifies favorable SNP alleles and candidate genes for frost tolerance in pea. BMC Genom. 2020, 21, 536. [Google Scholar] [CrossRef]
- Fransz, P.; Linc, G.; Lee, C.-R.; Aflitos, S.A.; Lasky, J.R.; Toomajian, C.; Ali, H.; Peters, J.; van Dam, P.; Ji, X.; et al. Molecular, genetic and evolutionary analysis of a paracentric inversion in Arabidopsis thaliana. Plant J. 2016, 88, 159–178. [Google Scholar] [CrossRef]
- Himmelbach, A.; Ruban, A.; Walde, I.; Šimková, H.; Doležel, J.; Hastie, A.; Stein, N.; Mascher, M. Discovery of multi-megabase polymorphic inversions by chromosome conformation capture sequencing in large-genome plant species. Plant J. 2018, 96, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Boideau, F.; Richard, G.; Coriton, O.; Huteau, V.; Belser, C.; Deniot, G.; Eber, F.; Falentin, C.; Ferreira de Carvalho, J.; Gilet, M.; et al. Epigenomic and structural events preclude recombination in Brassica napus. New Phytol. 2022, 234, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Baranger, A.; Aubert, G.; Arnau, G.; Lainé, A.L.; Deniot, G.; Potier, J.; Weinachter, C.; Lejeune-Hénaut, I.; Lallemand, J.; Burstin, J. Genetic diversity within Pisum sativum using protein- and PCR-based markers. Ther. Appl. Genet. 2004, 108, 1309–1321. [Google Scholar] [CrossRef]
- Conicella, C.; Errico, A. Karyotype Variations in Pisum sativum Ect. Abyssinicum. Caryologia 1990, 43, 87–97. [Google Scholar] [CrossRef]
- Trněný, O.; Brus, J.; Hradilová, I.; Rathore, A.; Das, R.R.; Kopecký, P.; Coyne, C.J.; Reeves, P.; Richards, C.; Smýkal, P. Molecular Evidence for Two Domestication Events in the Pea Crop. Genes 2018, 9, 535. [Google Scholar] [CrossRef]
- Fondevilla, S.; Satovic, Z.; Rubiales, D.; Moreno, M.T.; Torres, A.M. Mapping of quantitative trait loci for resistance to Mycosphaerella pinodes in Pisum sativum subsp. syriacum. Mol. Breed. 2008, 21, 439–454. [Google Scholar] [CrossRef]
- Tar’an, B.; Warkentin, T.; Somers, D.J.; Miranda, D.; Vandenberg, A.; Blade, S.; Woods, S.; Bing, D.; Xue, A.; DeKoeyer, D.; et al. Quantitative trait loci for lodging resistance, plant height and partial resistance to mycosphaerella blight in field pea (Pisum sativum L.). Ther. Appl. Genet. 2003, 107, 1482–1491. [Google Scholar] [CrossRef]
- Timmerman-Vaughan, G.M.; Moya, L.; Frew, T.J.; Murray, S.R.; Crowhurst, R. Ascochyta blight disease of pea (Pisum sativum L.): Defence-related candidate genes associated with QTL regions and identification of epistatic QTL. Ther. Appl. Genet. 2016, 129, 879–896. [Google Scholar] [CrossRef]
- Jha, A.B.; Gali, K.K.; Tar’an, B.; Warkentin, T.D. Fine Mapping of QTLs for Ascochyta Blight Resistance in Pea Using Heterogeneous Inbred Families. Front. Plant Sci. 2017, 8, 765. [Google Scholar] [CrossRef] [PubMed]
- Timmerman-Vaughan, G.M.; Frew, T.J.; Butler, R.; Murray, S.; Gilpin, M.; Falloon, K.; Johnston, P.; Lakeman, M.B.; Russell, A.; Khan, T. Validation of quantitative trait loci for Ascochyta blight resistance in pea (Pisum sativum L.), using populations from two crosses. Ther. Appl. Genet. 2004, 109, 1620–1631. [Google Scholar] [CrossRef]
- Zhu, H.; Gilchrist, L.; Hayes, P.; Kleinhofs, A.; Kudrna, D.; Liu, Z.; Prom, L.; Steffenson, B.; Toojinda, T.; Vivar, H. Does function follow form? Principal QTLs for Fusarium head blight (FHB) resistance are coincident with QTLs for inflorescence traits and plant height in a doubled-haploid population of barley. Ther. Appl. Genet. 1999, 99, 1221–1232. [Google Scholar] [CrossRef]
- Kolkman, J.M.; Kelly, J.D. QTL Conferring Resistance and Avoidance to White Mold in Common Bean. Crop Sci. 2003, 43, 539–548. [Google Scholar] [CrossRef]
- Aryamanesh, N.; Nelson, M.N.; Yan, G.; Clarke, H.J.; Siddique, K.H.M. Mapping a major gene for growth habit and QTLs for ascochyta blight resistance and flowering time in a population between chickpea and Cicer reticulatum. Euphytica 2010, 173, 307–319. [Google Scholar] [CrossRef]
- Lichtenzveig, J.; Bonfil, D.J.; Zhang, H.-B.; Shtienberg, D.; Abbo, S. Mapping quantitative trait loci in chickpea associated with time to flowering and resistance to Didymella rabiei the causal agent of Ascochyta blight. Ther. Appl. Genet. 2006, 113, 1357–1369. [Google Scholar] [CrossRef]
- Conner, R.; Hwang, S.-F.; Woods, S.; Chang, K.; Bing, D.; Su, H.; McAndrew, D.; Yager, L. Influence of agronomic traits on the expression of tissuespecific resistance to mycosphaerella blight in field pea. Can. J. Plant Sci. 2007, 87, 157–165. [Google Scholar] [CrossRef]
- Hamon, C.; Baranger, A.; Coyne, C.; McGee, R.; Goff, I.; L’Anthoëne, V.; Esnault, R.; Rivière, J.-P.; Klein, A.; Mangin, P.; et al. New consistent QTL in pea associated with partial resistance to Aphanomyces euteiches in multiple French and American environments. Ther. Appl. Genet. 2011, 123, 261–281. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Chang, K.; Hwang, S.; Gossen, B.; Howardand, R.; Warkentin, T. Components of disease resistance in desi and kabuli chickpea varieties against ascochyta blight. Plant Pathol. J. 2006, 5, 336–342. [Google Scholar] [CrossRef]
- Bradley, D.J.; Gilbert, G.S.; Parker, I.M. Susceptibility of clover species to fungal infection: The interaction of leaf surface traits and environment. Am. J. Bot. 2003, 90, 857–864. [Google Scholar] [CrossRef]
- Levitt, J. Responses of Plants to Environmental Stress. In Chilling, Freezing, and High Temperature Stresses; Academic Press: Cambridge, MA, USA, 1980; Volume 1. [Google Scholar]
- Jaškūnė, K.; Armonienė, R.; Liatukas, Ž.; Statkevičiūtė, G.; Cesevičienė, J.; Brazauskas, G. Relationship between Freezing Tolerance and Leaf Growth during Acclimation in Winter Wheat. Agronomy 2022, 12, 859. [Google Scholar] [CrossRef]
- Sack, L. Holding a Leaf Up to the Light. BioScience 2013, 63, 981–982. [Google Scholar]
- Baird, A.S.; Taylor, S.H.; Pasquet-Kok, J.; Vuong, C.; Zhang, Y.; Watcharamongkol, T.; Scoffoni, C.; Edwards, E.J.; Christin, P.-A.; Osborne, C.P.; et al. Developmental and biophysical determinants of grass leaf size worldwide. Nature 2021, 592, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Le May, C.; Ney, B.; Lemarchand, E.; Schoeny, A.; Tivoli, B. Effect of pea plant architecture on spatiotemporal epidemic development of ascochyta blight (Mycosphaerella pinodes) in the field. Plant Pathol. 2009, 58, 332–343. [Google Scholar] [CrossRef]
- Richard, B.; Bussière, F.; Langrume, C.; Rouault, F.; Jumel, S.; Faivre, R.; Tivoli, B. Effect of pea canopy architecture on microclimate and consequences on ascochyta blight infection under field conditions. Eur. J. Plant Pathol. 2013, 135, 509–524. [Google Scholar] [CrossRef]
- Miklas, P.N.; Porter, L.D.; Kelly, J.D.; Myers, J.R. Characterization of white mold disease avoidance in common bean. Eur. J. Plant Pathol. 2013, 135, 525–543. [Google Scholar] [CrossRef]
- Lester, D.R.; Ross, J.J.; Davies, P.J.; Reid, J.B. Mendel’ stem length gene (Le) encodes a gibberellin 3 β-hydroxylase. Plant Cell 1997, 9, 1435–1443. [Google Scholar] [PubMed]
- Achard, P.; Gong, F.; Cheminant, S.; Alioua, M.; Hedden, P.; Genschik, P. The Cold-Inducible CBF1 Factor–Dependent Signaling Pathway Modulates the Accumulation of the Growth-Repressing DELLA Proteins via Its Effect on Gibberellin Metabolism. Plant Cell 2008, 20, 2117–2129. [Google Scholar] [CrossRef]
- Saville, R.J.; Gosman, N.; Burt, C.J.; Makepeace, J.; Steed, A.; Corbitt, M.; Chandler, E.; Brown, J.K.M.; Boulton, M.I.; Nicholson, P. The ‘Green Revolution’ dwarfing genes play a role in disease resistance in Triticum aestivum and Hordeum vulgare. J. Exp. Bot. 2012, 63, 1271–1283. [Google Scholar] [CrossRef]
- Weller, J.L.; Liew, L.C.; Hecht, V.F.G.; Rajandran, V.; Laurie, R.E.; Ridge, S.; Wenden, B.; Vander Schoor, J.K.; Jaminon, O.; Blassiau, C.; et al. A conserved molecular basis for photoperiod adaptation in two temperate legumes. Proc. Natl. Acad. Sci. USA 2012, 109, 21158–21163. [Google Scholar] [CrossRef]
- Coyne, C.J.; Porter, L.D.; Boutet, G.; Ma, Y.; McGee, R.J.; Lesné, A.; Baranger, A.; Pilet-Nayel, M.-L. Confirmation of Fusarium root rot resistance QTL Fsp-Ps 2.1 of pea under controlled conditions. BMC Plant Biol. 2019, 19, 98. [Google Scholar] [CrossRef]
- Lejeune-Hénaut, I.; Bourion, V.; Etévé, G.; Cunot, E.; Delhaye, K.; Desmyter, C. Floral initiation in field-grown forage peas is delayed to a greater extent by short photoperiods, than in other types of European varieties. Euphytica 1999, 109, 201–211. [Google Scholar] [CrossRef]
- Roeber, V.M.; Bajaj, I.; Rohde, M.; Schmülling, T.; Cortleven, A. Light acts as a stressor and influences abiotic and biotic stress responses in plants. Plant Cell Environ. 2021, 44, 645–664. [Google Scholar] [CrossRef] [PubMed]
- Williams, O.; Vander Schoor, J.K.; Butler, J.B.; Ridge, S.; Sussmilch, F.C.; Hecht, V.F.G.; Weller, J.L. The genetic architecture of flowering time changes in pea from wild to crop. J. Exp. Bot. 2022, 73, 3978–3990. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| LGI | LGII | LGII | LGIV | LGV | LGVI | LGVII | Whole | |
|---|---|---|---|---|---|---|---|---|
| Number of markers | 271 | 382 | 572 | 396 | 348 | 373 | 402 | 2744 |
| Number of KASP SNPs | 174 | 235 | 405 | 268 | 255 | 222 | 253 | 1812 |
| Number of Markers/cM | 2.7 | 2.9 | 3.7 | 2.9 | 2.6 | 3.3 | 2.8 | 3.0 |
| Number of KASP SNPs/cM | 1.7 | 1.8 | 2.6 | 2.0 | 1.9 | 2.0 | 1.8 | 2.0 |
| Number of gaps > 2 cM between two contiguous markers | 9 | 8 | 11 | 17 | 13 | 8 | 13 | 79 |
| Number of gaps > 5 cM between two contiguous markers | 0 | 0 | 0 | 0 | 3 | 0 | 2 | 5 |
| Length (cM) | 101.8 | 131.4 | 155.8 | 137 | 136.4 | 111.5 | 141.3 | 915.2 |
| LG | MDA/MDAF QTL (Whole Analysis) | MDA/MDAF Peak (cM) | MDA/MDAF [CI] (cM) | MetaQTL (Analysis by Trait) | MetaQTL Peak (cM) | MetaQTL [CI] (cM) | Nb QTL/ChxTe | Nb QTL/JIxDP | Nb QTL/JIxFP | Parental Resistant Allele | R2 Max (%Var) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| LG I | MDA.1.1 | 90 | 3 | Dp.1.2 | 90 | 3 | 1 | 0 | 0 | Champagne | 27.5 |
| A.1 | 90 | 3 | StLe 1 | 0 | 0 | NA | 38.9 | ||||
| LG III | MDAF.3.1 | 152.1 | 0.9 | Dp.3.2 | 152.1 | 1.5 | 3 | 0 | 0 | Terese | 26.3 |
| A.3.1 | 152.2 | 1.1 | dflo 2 | * | * | NA | 1.6 | ||||
| NbBr 1 | NbBr 1 | 0 | 54.1 | ||||||||
| StLe 1 | 0 | 0 | 10.3 | ||||||||
| Ht 1 | Ht 1 | 0 | 91.6 | ||||||||
| NbNo 1 | NbNo 1 | 0 | 7.19 | ||||||||
| FR.3.1 | 152.1 | 1.6 | FRfd 2 | * | * | Terese | 7.4 | ||||
| MDAF.3.2 | 29.6 | 0.3 | Dp.3.3 | 29.8 | 1.4 | 3 | 5 | 0 | Champagne DP | 19 | |
| A.3.2 | 29.8 | 0.6 | dflo 3 | * | * | NA | 68.9 | ||||
| 0 | NbBr 1 | 0 | 19.3 | ||||||||
| FR.3.2 | 29.6 | 0.4 | FRfd 11 | * | * | Champagne | 60.2 | ||||
| LGV | MDAF.5.1 | 68.9 | 0.8 | Dp.5.1 | 68.9 | 0.9 | 6 | 1 | 6 | Champagne DP FP | 36.8 |
| A.5.1 | 65 | 4.6 | 0 | 0 | StLe 1 | NA | 23.5 | ||||
| A.5.2 | 69.1 | 2.5 | StLe 1 | StLe 1 | 0 | NA | 5.9 | ||||
| FR.5.1 | 67.7 | 2.3 | FRncc 1 | * | * | Champagne | 21.1 | ||||
| FRfd 3 | * | * | Champagne | 7.8 | |||||||
| MDAF.5.2 (MDAF.5.2.1 + MDAF.5.2.2) | 74.4 (72.0/76.6) | 1.7 (1.6/2.6) | Dp.5.2 | 72.5 | 2.1 | 4 | 2 | 2 | Champagne DP FP | 19.6 | |
| FR.5.2 | 71.8 | 3 | FRfd 4 | * | * | Champagne | 13.3 | ||||
| A.5.3 | 74.8 | 7 | NbBr 1 | 0 | 0 | NA | 6.3 | ||||
| Dp.5.3 | 77.4 | 3.1 | 0 | 5 | 0 | DP | 18.7 | ||||
| FR.5.3 | 78 | 1 | FRfd 1 | * | * | Champagne | 5.2 | ||||
| LGVI | MDA.6.1 | 34 | 3.2 | Dp.6.1 | 34 | 3.3 | 0 | 7 | 0 | DP | 29.4 |
| A.6.1 | 56.1 | 1.2 | 0 | StLe 1 | 0 | NA | 26.9 | ||||
| MDAF.6.2 | 56.9 | 0.7 | A.6.1 | 56.1 | 1.2 | dflo 3 | * | * | NA | 3.5 | |
| StLe 1 | 0 | 0 | 26.9 | ||||||||
| Dp.6.2 | 57.4 | 1 | 5 | 0 | 9 | Champagne FP | 35.1 | ||||
| FR.6.1 | 55.8 | 2.3 | FRfd 10 | * | * | Champagne | 14.4 | ||||
| A.6.2 | 59.1 | 2.6 | 0 | 0 | StLe 1 | NA | 28.6 | ||||
| NbBr 1 | 0 | 0 | 5.8 | ||||||||
| MDAF.6.3 | 64.4 | 2.5 | A.6.2 | 59.1 | 2.6 | 0 | NbBr 1 | 0 | NA | 5.8 | |
| FR.6.2 | 63.5 | 3.6 | FRncc 1 | * | * | Champagne | 14.5 | ||||
| Dp.6.3 | 65.3 | 3.6 | 2 | 0 | 0 | Champagne | 29.3 | ||||
| LGVII | MDA.7.1 | 68.3 | 1.3 | Dp.7.1 | 68.3 | 1.3 | 0 | 2 | 0 | DP | 35 |
| 1.3 | A.7.1 | 68.3 | 6.7 | 0 | NbBr 1 | 0 | NA | 11.2 | |||
| MDA.7.2 | 81.2 | 12.7 | Dp.7.2 | 93 | 13.6 | 0 | 0 | 1 | FP | 13.9 | |
| 12.7 | A.7.2 | 81.2 | 12.7 | 0 | 0 | StLe 1 | NA | 14.6 |
| Increase in D. pinodes Resistance and Frost Tolerance | ||||||
|---|---|---|---|---|---|---|
| Increase in | Number of branches | MDAF.3.1 | MDAF.6.2 | MDAF.3.2 | MDAF.5.2 | |
| Delay in | Flowering date | MDAF.3.1 | MDAF.6.2 | MDAF.3.2 | ||
| Decrease in | Stipule length | MDAF.3.1 | MDAF.6.2 | MDAF.5.1 | ||
| Decrease in | Plant height | MDAF.3.1 | ||||
| Decrease in | Number of nodes | MDAF.3.1 | ||||
| Population(s) | GWAS | China x Cameor | Champagne x Terese | Champagne x Terese | JI296 x FP | JI296 x DP | JI296 x DP | P665 x Messire | P665 x Messire | A88 x Rovar | A26 x Rovar | A88 x Rovar/A26 x Rovar | Carneval x MP1401 | P651 (P.fulvum) x Alfetta |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Disease assessment conditions | Controlled, inoculated, seedlings, | Controlled, inoculated, seedlings, | Controlled, inoculated, seedlings, | Controlled, Field inoculated, seedlings, adult plants | Controlled, Field inoculated, seedlings, adult plants | Controlled, Field inoculated, seedlings, adult plants | Field, natural epidemics, adult plants | Field, natural epidemics, adult plants | Field, natural epidemics, adult plants | Field, natural epidemics, adult plants | Field, natural epidemics, adult plants ** + Controlled, inoculated, seedlings *** | |||
| Frost assessment conditions | Field and Controlled | Field | Field and Controlled | Field and Controlled | ||||||||||
| LG I | (WFD 1.1) | Asc1.1 | Asc1.1 | abI-IV-1 ** | ||||||||||
| LDBlock I.1 | ||||||||||||||
| (Dp.1.1) | ||||||||||||||
| (Dp.1.2) | ||||||||||||||
| (Dp.1.3 *) | ||||||||||||||
| LG II | mpII-1, mpII-2 | MpII.1 | MpII.1 | (Asc2.1) | Asc2.1, Asc2.2 | Asc2.1 | MbII | |||||||
| LG III | III.1 | Dp.3.1 | MpIII.1 | Asc3.2 ? | Asc3.2 ? | abIII-2 *** | ||||||||
| LDBlock III.1 | III.1 | WFD 3.2 * | FR.3.1 */Dp.3.2 * | |||||||||||
| MpIII.1 | ||||||||||||||
| (Dp.3.4) | mpIII-1 | |||||||||||||
| (Dp.3.5 *) | ||||||||||||||
| (Dp.3.6 *) | MpIII.2 | |||||||||||||
| (Dp.3.7) | mpIII.4 ? | MpIII.4 | ||||||||||||
| (Dp.3.8) | mpIII-5 | |||||||||||||
| WFD 3.1 | FR.3.2/Dp.3.3 | Dp.3.3 | mpIII-3 | MpIII.3 | MpIII.3 | Asc3.1 ? | Asc3.1 ? | Asc3.1 ? | abIII-1 ** | |||||
| Dp.3.9 | ||||||||||||||
| mpIII-2 | MpIII.5 ? | |||||||||||||
| LG IV | MpIV.1 | MpIV.1 | Asc4.1, Asc4.2, Asc4.3 | Asc4.1 | Asc4.2, Asc4.3 | MbIV | abI-IV-2 **, abI-IV-3 **, abI-IV-4 **, abI-IV-5 *** | |||||||
| LG V | V.1 | |||||||||||||
| LDBlock V.1 | WFD 5.1 | |||||||||||||
| FR.5.1/Dp.5.1 | Dp.5.1 | Dp.5.1 | mpVa-1 | Mp.V.1 | MpV.3 | Asc5.1 | Asc5.1 | |||||||
| FR.5.2/Dp.5.2 | Dp.5.2 | Dp.5.2 | ||||||||||||
| Dp.5.3 | MpV.2 | |||||||||||||
| V.2 | VpV.1 | |||||||||||||
| (WFD 5.2) | (FR.5.3) | |||||||||||||
| LG VI | ||||||||||||||
| LDBlock VI.1/LDBlock VI.2 | VI.1 | WFD 6.1 | FR.6.1/Dp.6.2 | Dp.6.2 | Dp.6.1 | mpVI-1 | mp.VI.1 | mp.VI.1 | MbVI | |||||
| FR.6.2/Dp.6.3 | ||||||||||||||
| (Dp.6.4 *) | ||||||||||||||
| LG VII | VII.2 | Asc7.1 | Asc7.3, Asc7.1, Asc7.2 | abVII-1 **, abVII-2 *** | ||||||||||
| LDBlock VII.1 | ||||||||||||||
| Dp.7.1 | mpVII-1 | |||||||||||||
| (Dp.7.2) | ||||||||||||||
| Dp.7.3 | ||||||||||||||
| LG I | Beji et al. 2020 [52] | Klein et al. 2014 [29] | Lejeune et al. 2008 [30] /Dumont et al. 2009 [31] | This study | Prioul et al. 2004 [24] | Fondevilla et al. 2008 [59] / Fondevilla et al. 2011 [27] | Carrillo et al. 2014 [28] | Timmerman-Vaughan et al. 2002 [23] | Timmerman-Vaughan et al. 2004 [63] | Timmerman-Vaughan et al. 2016 [61] | Ta’ran et al. 2003 [60] | Jha et al. 2016 [26] | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boutet, G.; Lavaud, C.; Lesné, A.; Miteul, H.; Pilet-Nayel, M.-L.; Andrivon, D.; Lejeune-Hénaut, I.; Baranger, A. Five Regions of the Pea Genome Co-Control Partial Resistance to D. pinodes, Tolerance to Frost, and Some Architectural or Phenological Traits. Genes 2023, 14, 1399. https://doi.org/10.3390/genes14071399
Boutet G, Lavaud C, Lesné A, Miteul H, Pilet-Nayel M-L, Andrivon D, Lejeune-Hénaut I, Baranger A. Five Regions of the Pea Genome Co-Control Partial Resistance to D. pinodes, Tolerance to Frost, and Some Architectural or Phenological Traits. Genes. 2023; 14(7):1399. https://doi.org/10.3390/genes14071399
Chicago/Turabian StyleBoutet, Gilles, Clément Lavaud, Angélique Lesné, Henri Miteul, Marie-Laure Pilet-Nayel, Didier Andrivon, Isabelle Lejeune-Hénaut, and Alain Baranger. 2023. "Five Regions of the Pea Genome Co-Control Partial Resistance to D. pinodes, Tolerance to Frost, and Some Architectural or Phenological Traits" Genes 14, no. 7: 1399. https://doi.org/10.3390/genes14071399
APA StyleBoutet, G., Lavaud, C., Lesné, A., Miteul, H., Pilet-Nayel, M.-L., Andrivon, D., Lejeune-Hénaut, I., & Baranger, A. (2023). Five Regions of the Pea Genome Co-Control Partial Resistance to D. pinodes, Tolerance to Frost, and Some Architectural or Phenological Traits. Genes, 14(7), 1399. https://doi.org/10.3390/genes14071399