
Transcriptome Characterization and Gene Changes Induced by Fusarium solani in Sweetpotato Roots
Abstract
1. Introduction
2. Materials and Methods
2.1. Sweetpotato and Fungal Materials
2.2. Inoculation of F. solani
2.3. RNA Extraction, Library Preparation, and Illumina Sequencing
2.4. Data Analysis
2.5. Validations of RNA-seq Data Using Quantitative Real-Time Polymerase Chain Reaction
3. Results
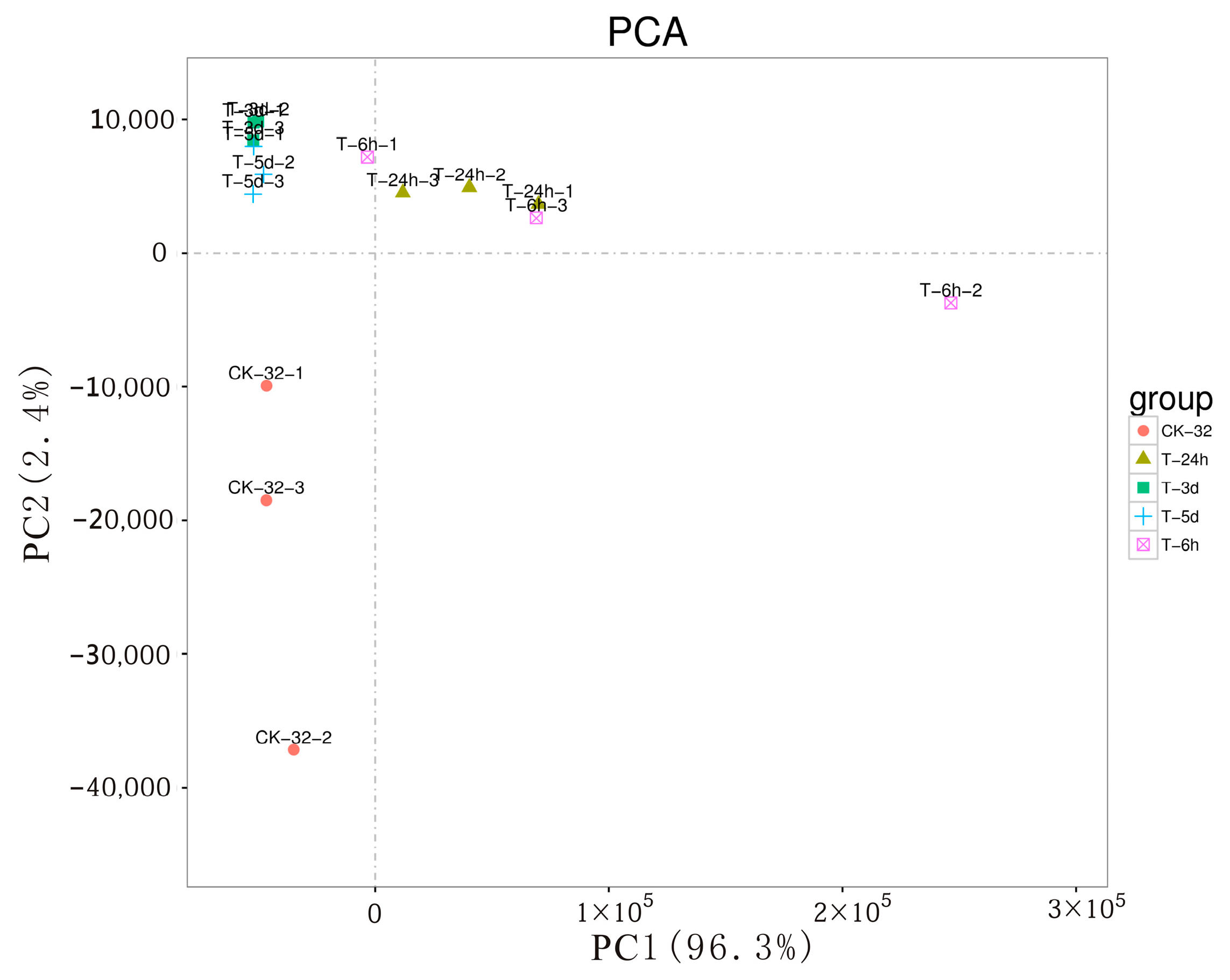
3.1. Disease Severity, Transcriptome Generation, and Assembly
3.2. Analysis of Sweetpotato DEGs in Response to F. solani
3.3. Verification of the RNA-seq Results
3.4. GO and KEGG Analyses of the DEGs
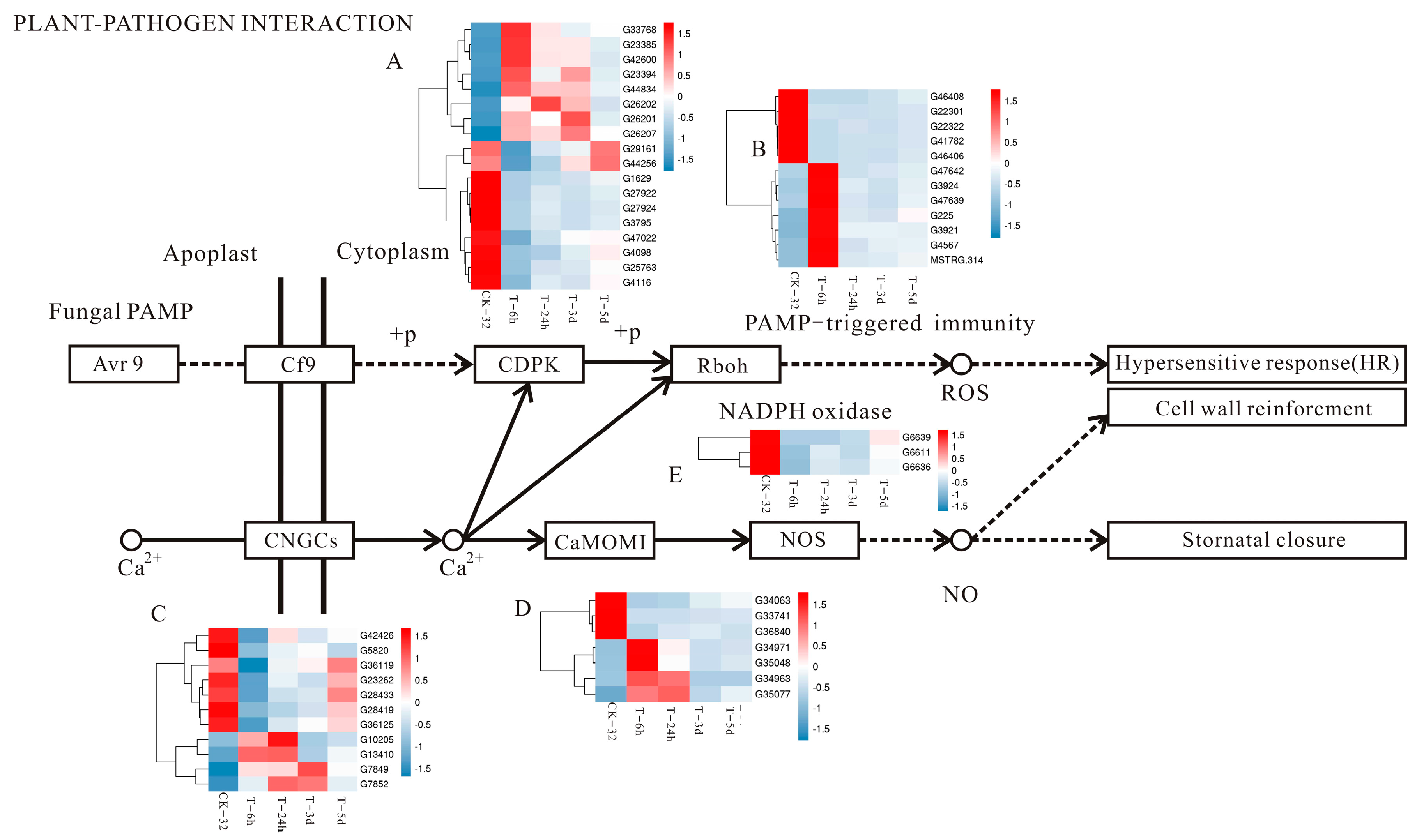
3.5. Plant–Pathogen Interaction
3.6. Expression of TFs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zang, N.; Zhai, H.; Gao, S.; Chen, W.; He, S.; Liu, Q. Efficient production of transgenic plants using the bar gene for herbicide resistance in sweetpotato. Sci. Hortic. 2009, 122, 649–653. [Google Scholar] [CrossRef]
- Munoz-Rodriguez, P.; Carruthers, T.; Wood, J.R.I.; Williams, B.R.M.; Weitemier, K.; Kronmiller, B.; Ellis, D.; Anglin, N.L.; Longway, L.; Harris, S.A.; et al. Reconciling conflicting phylogenies in the origin of sweetpotato and dispersal to polynesia. Curr. Biol. 2018, 28, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Gao, B.; Li, X.; Ma, J.; Chen, S. First report of Fusarium solani causing Fusarium root rot and stem canker on storage roots of sweetpotato in china. Plant Dis. 2014, 98, 160. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Ma, T.; Zhao, N.; Zhang, X.; Fang, B.; Huang, L. Whole-genome sequencing and comparative genome analysis of Fusarium solani-melongenae causing Fusarium root and stem rot in sweetpotatoes. Microbiol. Spectr. 2022, 10, e0068322. [Google Scholar] [CrossRef]
- Jassim, S.J.; Al-Salami, I.; Al-Shujairi, K.A. Molecular diagnosis of some isolates of Fusarium solani isolated from potato tubers (Solanum tuberosum L.). Plant Pathol. 2008, 57, 832–862. [Google Scholar]
- Batnini, M.; Haddoudi, I.; Taamali, W.; Djebali, N.; Badri, M.; Mrabet, M.; Mhadhbi, H. Medicago truncatula in interaction with Fusarium and Rhizoctonia phytopathogenic fungi: Fungal aggressiveness, plant response biodiversity and character heritability indices. Plant Pathol. J. 2021, 37, 315–328. [Google Scholar] [CrossRef]
- Rostami, A.; Sadravi, M.; Rezaee, R.; Abdollahi, M. Biological control of Fusarium root rot of bean with two Trichoderma species and Pseudomonas fluorescens. Plant Pathol. Sci. 2020, 9, 14–27. [Google Scholar] [CrossRef]
- Huang, L.; Fang, B.; Chen, J.; Zhang, X.; Luo, Z.; Yang, Y.; Yao, Z.; Chen, X. Sweetpotato Disease Identification and Control Guide; China Agriculture Press: Beijing, China, 2020. [Google Scholar]
- Yuan, F.; Rasheed, A.; Zhu, Z.; Wulff, B.B.H.; He, Z. Harnessing wheat fhb1 for Fusarium resistance. Trends Plant Sci. 2020, 25, 1–3. [Google Scholar]
- Wang, H.; Sun, S.; Ge, W.; Zhao, L.; Hou, B.; Wang, K.; Lyu, Z.F.; Chen, L.; Xu, S.; Guo, J.; et al. Horizontal gene transfer of fhb7 from fungus underlies Fusarium head blight resistance in wheat. Science 2020, 368, 5435. [Google Scholar] [CrossRef]
- Kage, U.; Yogendra, K.N.; Kushalappa, A.C. TaWRKY70 transcription factor in wheat QTL-2DL regulates downstream metabolite biosynthetic genes to resist Fusarium graminearum infection spread within spike. Sci. Rep. 2017, 15, 42596. [Google Scholar] [CrossRef]
- Wang, C.; He, X.; Li, Y.; Wang, L.; Guo, X.; Guo, X. The cotton MAPK kinase GhMPK20 negatively regulates resistance to Fusarium oxysporum by mediating the MKK4-MPK20-WRKY40 cascade. Mol. Plant Pathol. 2018, 19, 1624–1638. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.J.; Lu, X.S.; Chen, Q.; Zhang, M.J.; Qu, Y.Y. Effect of silencing GbGSTU7 gene on the resistance to Fusarium Wilt of Gossypium barbadense. Chin. J. Agric. Biotechnol. 2022, 30, 434–441. [Google Scholar]
- Man, M.; Zhu, Y.; Liu, L.; Luo, L.; Han, X.; Qiu, L.; Xing, Y. Defense mechanisms of cotton Fusarium and Verticillium wilt and comparison of pathogenic response in cotton and humans. Int. J. Mol. Sci. 2022, 23, 12217. [Google Scholar] [CrossRef]
- Cilkiz, M. Genome-Wide Bioinformatic and Functional Analysis of Receptor-like Proteins in Cotton. Master’s Thesis, Texas A & M University, College Station, TX, USA, 2017. [Google Scholar]
- Scott, J.; Agrama, H.; Jones, J. RFLP-based analysis of recombination among resistance genes to Fusarium wilt races 1, 2, and 3 in tomato. J. Am. Soc. Hortic. Sci. 2004, 129, 394–400. [Google Scholar] [CrossRef]
- Bournival, B.L.; Scott, J.W.; Vallejos, C.E. An isozyme marker for resistance to race-3 of Fusarium oxysporum f. sp. Lycopersici in tomato. Theor. Appl. Genet. 1989, 78, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Chitwood-Brown, J.; Vallad, G.E.; Lee, T.G.; Hutton, S.F. Breeding for resistance to Fusarium wilt of tomato: A review. Genes 2021, 12, 1673. [Google Scholar] [CrossRef]
- Liu, E.; Li, Z.; Luo, Z.; Xu, L.; Jin, P.; Ji, S.; Zhou, G.; Wang, Z.; Zhou, Z.; Zhang, H. Genome-wide identification of DUF668 gene family and expression analysis under drought and salt stresses in sweetpotato [Ipomoea batatas (L.) Lam]. Genes 2023, 14, 217. [Google Scholar] [CrossRef]
- Wang, C.; Wang, L.; Lei, J.; Chai, S.; Jin, X.; Zou, Y.; Sun, X.; Mei, Y.; Cheng, X.; Yang, X.; et al. IbMYB308, a sweetpotato R2R3-MYB gene, improves salt stress tolerance in transgenic tobacco. Genes 2022, 13, 1476. [Google Scholar] [CrossRef]
- Jiang, W.; Jin, R.; Wang, D.; Yang, Y.; Zhao, P.; Liu, M.; Zhang, A.; Tang, Z. A novel high-affinity potassium transporter IbHKT-like gene enhances low-potassium tolerance in transgenic roots of sweetpotato (Ipomoea batatas (L.) Lam.). Plants 2022, 11, 1389. [Google Scholar] [CrossRef]
- Zhu, H.; Yang, X.; Wang, X.; Li, Q.; Guo, J.; Ma, T.; Zhao, C.; Tang, Y.; Qiao, L.; Wang, J.; et al. The sweetpotato β-amylase gene IbBAM1.1 enhances drought and salt stress resistance by regulating ROS homeostasis and osmotic balance. Plant Physiol. Biochem. 2021, 168, 167–176. [Google Scholar] [CrossRef]
- Nie, N.; Huo, J.; Sun, S.; Zuo, Z.; Chen, Y.; Liu, Q.; Zhai, H. Genome-wide characterization of the PIFs family in sweetpotato and functional identification of IbPIF3.1 under drought and Fusarium wilt stresses. Int. J. Mol. Sci. 2023, 24, 4092. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, Q.; Zhai, H.; Gao, S.; Yang, L.; Wang, Z.; He, S. IbBBX24 promotes the jasmonic acid pathway and enhances Fusarium wilt resistance in sweetpotato. Plant Cell 2020, 32, 1102–1123. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Zhang, H.; Zhang, Q.; Zhai, H.; Liu, Q.; He, S. The plasma membrane-localized sucrose transporter IbSWEET10 contributes to the resistance of sweetpotato to Fusarium oxysporum. Front. Plant Sci. 2017, 8, 197. [Google Scholar] [PubMed]
- Arisha, M.H.; Aboelnasr, H.; Ahmad, M.Q.; Liu, Y.; Tang, W.; Gao, R.; Yan, H.; Kou, M.; Wang, X.; Zhang, Y.; et al. Transcriptome sequencing and whole genome expression profiling of hexaploid sweetpotato under salt stress. BMC Genom. 2020, 21, 197. [Google Scholar] [CrossRef]
- Soviguidi, D.R.J.; Liu, Y.; Pan, R.; Abou-Elwafa, S.F.; Rao, L.; Abel, S.; Zhang, W.; Yang, X. Role of sweetpotato GST genes in abiotic stress tolerance revealed by genomic and transcriptomic analyses. Crop Breed. Appl. Biotechnol. 2022, 22, e36852212. [Google Scholar] [CrossRef]
- Lin, Y.; Zou, W.; Lin, S.; Onofua, D.; Yang, Z.; Chen, H.; Chen, X. Transcriptome profiling and digital gene expression analysis of sweetpotato for the identification of putative genes involved in the defense response against Fusarium oxysporum f. sp. batatas. PLoS ONE 2017, 12, e0187838. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Bigeard, J.; Colcombet, J.; Hirt, H. Signaling mechanisms in pattern-triggered immunity (PTI). Mol. Plant 2015, 8, 521–539. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.G.; Dang, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Riechmann, J.L.; Ratcliffe, O.J. A genomic perspective on plant transcription factors. Curr. Opin. Plant Biol. 2000, 3, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.A.; Moyer, J.W.; Ferri, D.M.; Smith, T.P.; Holmes, G.J. Fusarium root rot and stem canker and surface rot. In Compendium of Sweetpotato Diseases, Pests, and Disorders, 2nd ed.; The American Phytopathological Society: St. Paul, MN, USA, 2013; pp. 37–40. [Google Scholar]
- Jiang, N.; Fan, X.; Lin, W.; Wang, G.; Cai, K. Transcriptome analysis reveals new insights into the bacterial wilt resistance mechanism mediated by silicon in tomato. Int. J. Mol. Sci. 2019, 20, 761. [Google Scholar] [CrossRef] [PubMed]
- Javaid, M.H.; Khan, A.R.; Salam, A.; Neelam, A.; Azhar, W.; Ulhassan, Z.; Gan, Y. Exploring the adaptive responses of plants to abiotic stresses using transcriptome data. Agriculture 2022, 12, 211. [Google Scholar] [CrossRef]
- Sunkar, R. Plant stress tolerance. Methods Mol. Biol. 2010, 639, 401. [Google Scholar]
- Li, Y.; Yin, Y.; Golding, J.B.; Geng, S.; Chen, G.; Yang, H. Metabolomic and transcriptomic analyses of quality deterioration in Fusarium solani infected sweetpotato (Ipomoea batatas (L.) Lam cv Xinxiang) storage roots. J. Agric. Food Chem. 2022, 70, 7258–7266. [Google Scholar] [CrossRef]
- Duan, Y.; Ma, S.; Chen, X.; Shen, X.; Yin, C.; Mao, Z. Transcriptome changes associated with apple (Malus domestica) root defense response after Fusarium proliferatum f. sp. malus domestica infection. BMC Genom. 2022, 23, 484. [Google Scholar] [CrossRef]
- Camejo, D.; Guzmán-Cedeño, Á.; Moreno, A. Reactive oxygen species, essential molecules, during plant-pathogen interactions. Plant Physiol. Biochem. 2016, 103, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Waheed, A.; Haxim, Y.; Islam, W.; Kahar, G.; Liu, X.; Zhang, D. Role of pathogen’s effectors in understanding host-pathogen interaction. Biochim. Biophys. Acta-Mol. Cell Res. 2022, 1869, 119347. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, C. Pattern-recognition receptors in plant innate immunity. Curr. Opin. Immunol. 2008, 20, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Rushton, P.J.; Torres, J.T.; Parniske, M.; Wernert, P.; Hahlbrock, K.; Somssich, I.E. Interaction of elicitor-induced DNA-binding proteins with elicitor response elements in the promoters of parsley PR1 genes. EMBO J. 1996, 15, 5690–5700. [Google Scholar] [CrossRef] [PubMed]
- Rushton, P.J.; Somssich, I.E.; Ringler, P.; Shen, Q.J. WRKY transcription factors. Trends Plant Sci. 2010, 15, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef]
- Jin, R.; Kim, H.S.; Yu, T.; Zhang, A.; Yang, Y.; Liu, M.; Tang, Z. Identification and function analysis of bHLH genes in response to cold stress in sweetpotato. Plant Physiol. Biochem. 2021, 169, 224–235. [Google Scholar] [CrossRef]
- Toledo-Ortiz, G.; Huq, E.; Quail, P.H. The Arabidopsis basic/helix-loop-helix transcription factor family. Plant Cell 2003, 15, 1749–1770. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5′–3′) | Name | Sequence (5′–3′) |
|---|---|---|---|
| Actin-F | AGCAGCATGAAGATTAAGGTTGTAGCAC | Actin-R | TGGAAAATTAGAAGCACTTCCTGTGAAC |
| qG38170-F | TCCAACTCTGAGCCGCCGCAGC | qG38170-R | TCCAAACTCTCCCCAATTAT |
| qG43500-F | ATGCAGGGGGCCATAAAACT | qG43500-R | ACTCAATAGCGCCTCCATCC |
| qG42363-F | CAAGGTCCCCGGAGGATGTAACA | qG42363-R | CTGAGGATAACTATAAGCATCAG |
| qG15761-F | GCCATCCTGAACCGGAGCGTCA | qG15761-R | CTGGTAAACAAGCTCCACCGTC |
| qG13937-F | TGTTTTGCTTGTGGTGGTGGCGC | qG13937-R | ACAGAAGGCAGTCCACCCATACT |
| Comparisons | Biological Process | Up | Down | Molecular Function | Up | Down | Cellular Component | Up | Down |
|---|---|---|---|---|---|---|---|---|---|
| CK-32 vs. T-6h | metabolic process | 613 | 1831 | catalytic activity | 635 | 1584 | cell part | 269 | 952 |
| cellular process | 546 | 1645 | binding | 440 | 1213 | cell | 269 | 952 | |
| single-organism process | 508 | 1303 | transporter activity | 81 | 161 | organelle | 182 | 731 | |
| all | 3022/9230 | all | 2920/8826 | all | 1722/5613 | ||||
| CK-32 vs.T-24h | metabolic process | 748 | 1051 | catalytic activity | 789 | 916 | cell part | 354 | 545 |
| single-organism process | 588 | 787 | binding | 493 | 755 | cell | 354 | 545 | |
| cellular process | 641 | 999 | transporter activity | 87 | 115 | organelle | 244 | 437 | |
| all | 2253/9230 | all | 2233/8826 | all | 1301/5613 | ||||
| CK-32 vs. T-3d | cellular process | 756 | 1381 | catalytic activity | 935 | 1260 | cell part | 370 | 763 |
| metabolic process | 869 | 1491 | binding | 576 | 998 | cell | 370 | 763 | |
| single-organism process | 711 | 1089 | transporter activity | 111 | 140 | organelle | 259 | 610 | |
| all | 2906/9230 | all | 1669/5613 | all | 2840/8826 | ||||
| CK-32 vs. T-5d | single-organism process | 707 | 702 | catalytic activity | 927 | 772 | cell part | 370 | 466 |
| cellular process | 722 | 910 | binding | 536 | 627 | cell | 370 | 466 | |
| metabolic process | 853 | 960 | transporter activity | 115 | 98 | organelle | 267 | 379 | |
| all | 2236/9230 | all | 1249/5613 | all | 2195/8826 | ||||
| Comparisons | Pathway | CGPA (% of 12,225) | The Top 3 (DEGs Number/All Genes Number) | ||
|---|---|---|---|---|---|
| CK-32 vs. T-6h | 130 | 3728 (30.49%) | Metabolic pathways (1800/5181) | Biosynthesis of secondary metabolites (1207/3087) | Carbon metabolism (267/683) |
| CK-32 vs. T-24h | 130 | 2766 (22.63%) | Metabolic pathways (1391/5171) | Biosynthesis of secondary metabolites (945/3087) | Carbon metabolism (223/683) |
| CK-32 vs. T-3d | 129 | 3562 (29.14%) | Metabolic pathways (1843/5171) | Biosynthesis of secondary metabolites (1229/3087) | Carbon metabolism (282/683) |
| CK-32 vs. T-5d | 129 | 2732 (22.35%) | Metabolic pathways (1493/5171) | Biosynthesis of secondary metabolites (1041/3087) | Carbon metabolism (240/683) |
| Transcription Factor | CK-32 vs. T-6h | CK-32 vs. T-24h | CK-32 vs. T-3d | CK-32 vs. T-5d |
|---|---|---|---|---|
| AP2 | 2 | 2 | 4 | 3 |
| ARF | 8 | 6 | 8 | 7 |
| ARR-B | 3 | 1 | 1 | 1 |
| B3 | 1 | 2 | 2 | 1 |
| BBR | 1 | 1 | 1 | 1 |
| BES1 | 3 | 2 | 3 | 4 |
| bHLH | 29 | 23 | 32 | 27 |
| bZIP | 7 | 8 | 9 | 6 |
| C2H2 | 16 | 11 | 11 | 10 |
| C3H | 8 | 3 | 3 | 2 |
| CAMTA | 0 | 1 | 0 | 0 |
| CO-like | 1 | 0 | 0 | 0 |
| CPP | 1 | 1 | 1 | 0 |
| DBB | 3 | 0 | 3 | 0 |
| Dof | 7 | 3 | 7 | 4 |
| E2F/DP | 2 | 1 | 1 | 2 |
| ERF | 5 | 4 | 4 | 5 |
| FAR1 | 3 | 0 | 1 | 0 |
| G2-like | 9 | 6 | 11 | 10 |
| GATA | 5 | 2 | 6 | 5 |
| GRAS | 4 | 1 | 1 | 1 |
| GRF | 0 | 0 | 1 | 0 |
| HB-other | 6 | 3 | 2 | 1 |
| HD-ZIP | 15 | 10 | 13 | 10 |
| HSF | 8 | 7 | 7 | 7 |
| LBD | 1 | 1 | 4 | 3 |
| M-type | 1 | 0 | 1 | 0 |
| MIKC | 0 | 1 | 2 | 2 |
| MYB | 22 | 20 | 28 | 25 |
| NAC | 18 | 17 | 20 | 17 |
| NF | 4 | 3 | 3 | 3 |
| Nin-like | 0 | 0 | 1 | 0 |
| SBP | 5 | 3 | 4 | 3 |
| STAT | 0 | 0 | 1 | 0 |
| TALE | 7 | 6 | 6 | 5 |
| TCP | 4 | 1 | 2 | 2 |
| Trihlix | 4 | 2 | 3 | 2 |
| Whrly | 0 | 0 | 1 | 0 |
| WRKY | 17 | 21 | 24 | 17 |
| Upregulated | 35/230 | 45/174 | 45/232 | 43/188 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Luo, Q.; Tang, W.; Ma, J.; Yang, D.; Chen, J.; Gao, F.; Sun, H.; Xie, Y. Transcriptome Characterization and Gene Changes Induced by Fusarium solani in Sweetpotato Roots. Genes 2023, 14, 969. https://doi.org/10.3390/genes14050969
Zhang C, Luo Q, Tang W, Ma J, Yang D, Chen J, Gao F, Sun H, Xie Y. Transcriptome Characterization and Gene Changes Induced by Fusarium solani in Sweetpotato Roots. Genes. 2023; 14(5):969. https://doi.org/10.3390/genes14050969
Chicago/Turabian StyleZhang, Chengling, Qinchuan Luo, Wei Tang, Jukui Ma, Dongjing Yang, Jingwei Chen, Fangyuan Gao, Houjun Sun, and Yiping Xie. 2023. "Transcriptome Characterization and Gene Changes Induced by Fusarium solani in Sweetpotato Roots" Genes 14, no. 5: 969. https://doi.org/10.3390/genes14050969
APA StyleZhang, C., Luo, Q., Tang, W., Ma, J., Yang, D., Chen, J., Gao, F., Sun, H., & Xie, Y. (2023). Transcriptome Characterization and Gene Changes Induced by Fusarium solani in Sweetpotato Roots. Genes, 14(5), 969. https://doi.org/10.3390/genes14050969