
A New Assessment of Robust Capuchin Monkey (Sapajus) Evolutionary History Using Genome-Wide SNP Marker Data and a Bayesian Approach to Species Delimitation

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
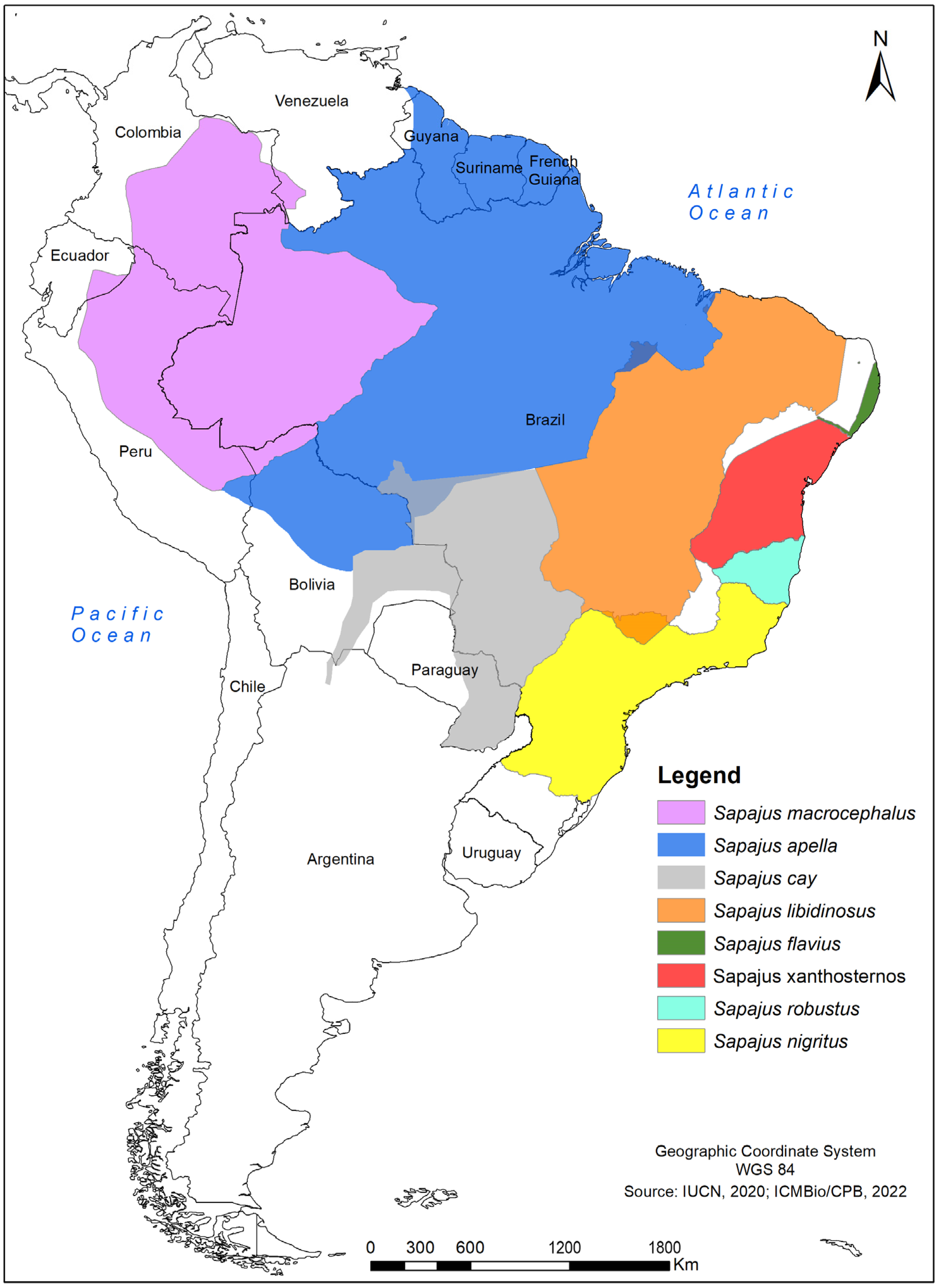
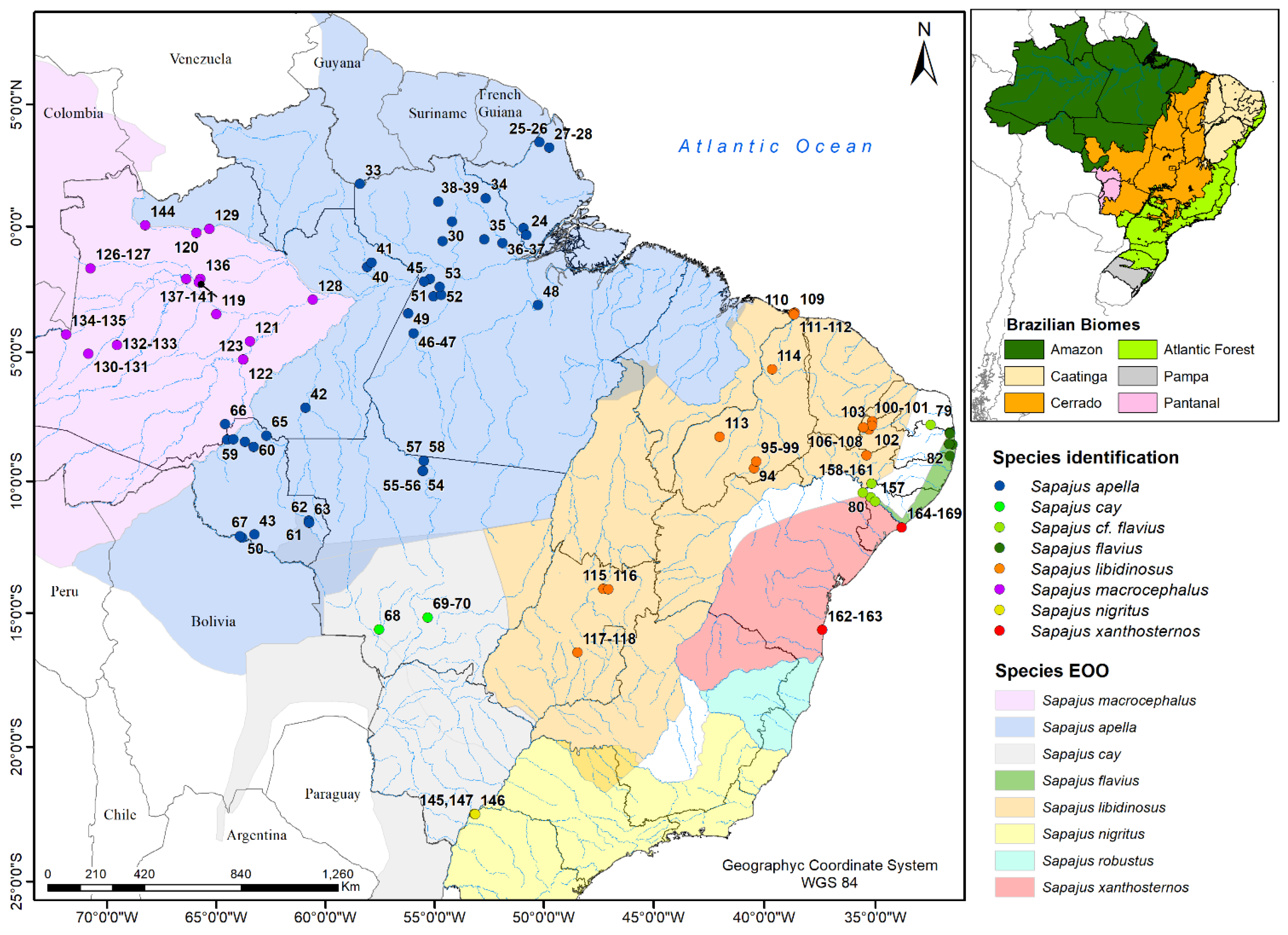
2.1. Samples
2.2. DNA Extraction and Quantification
2.3. Whole Genome Markers
2.4. Quality Control
2.5. Assembly and SNP Calling
2.6. Phylogenetic Analysis
2.7. Bayesian Analysis and Divergence Dating
3. Results
3.1. Molecular Data
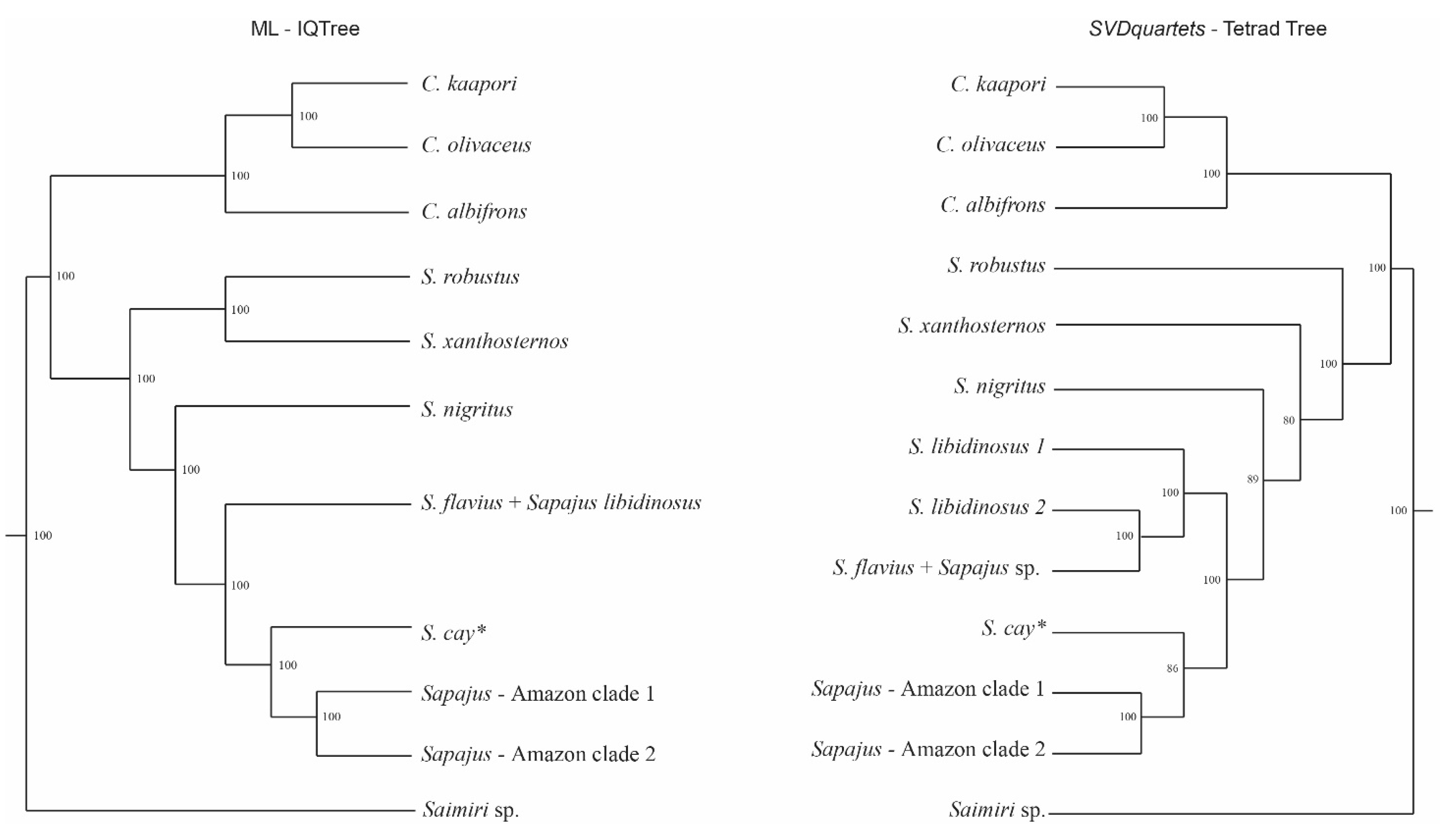
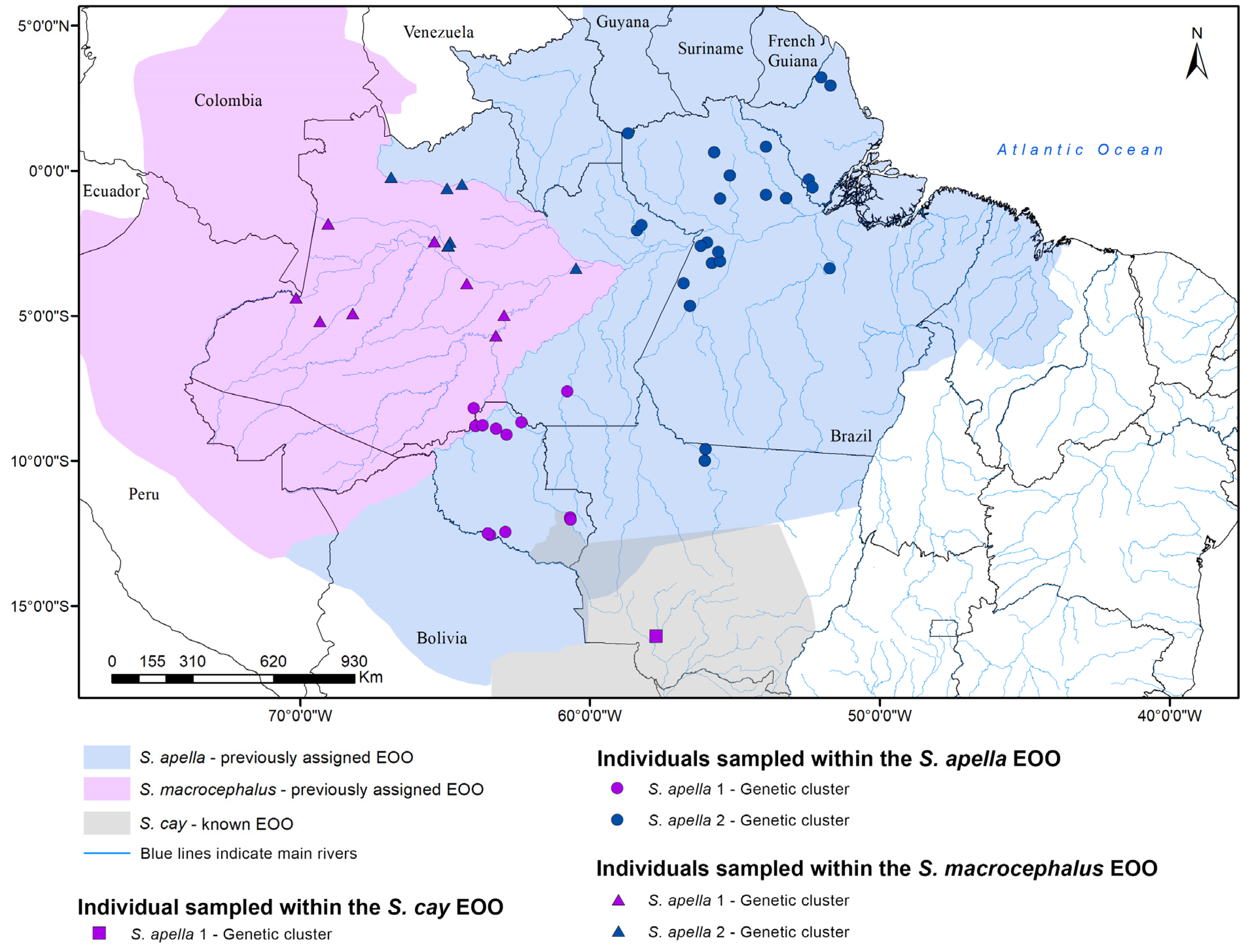
3.2. Sapajus Phylogenetic Reconstructions
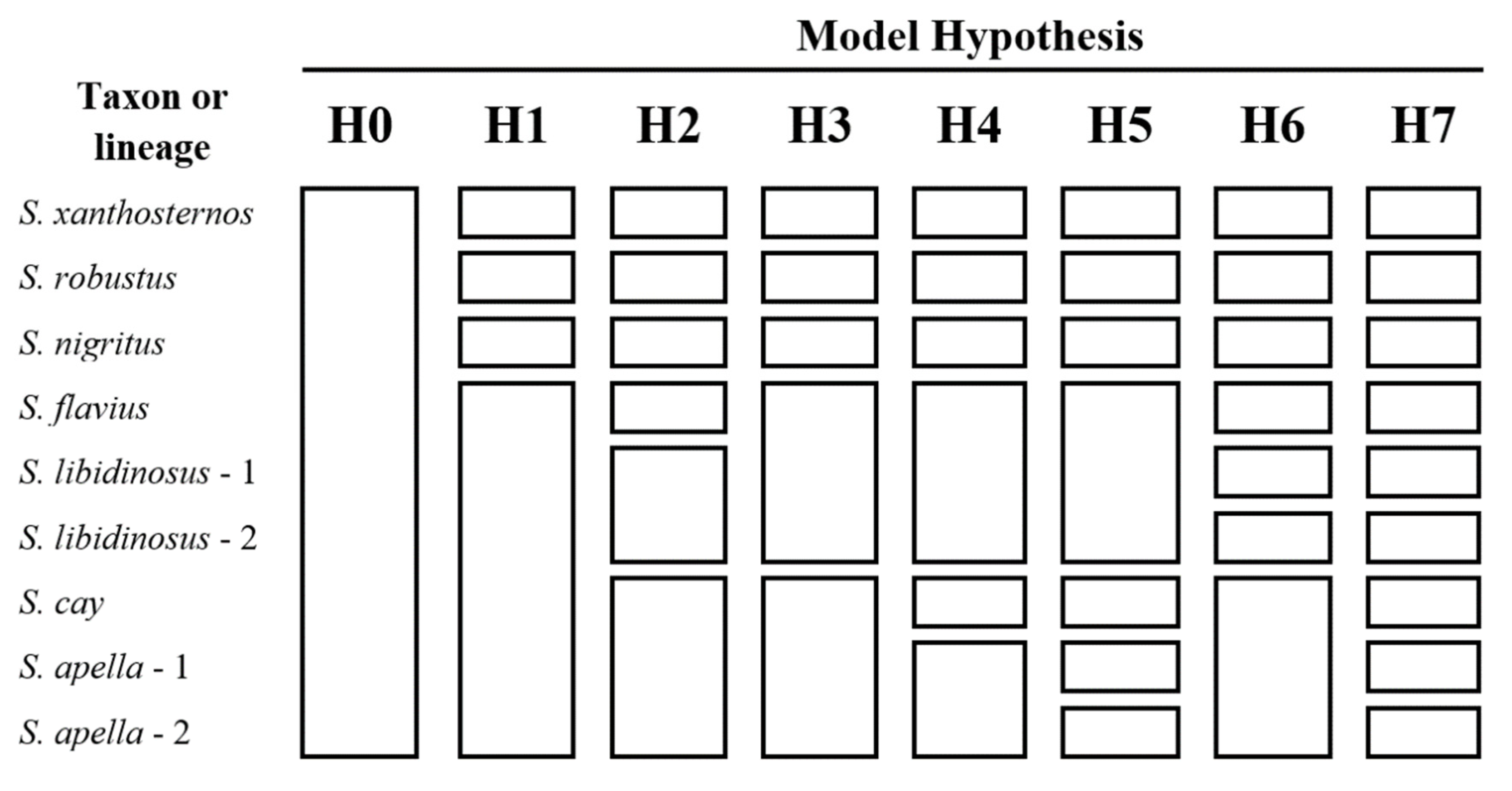
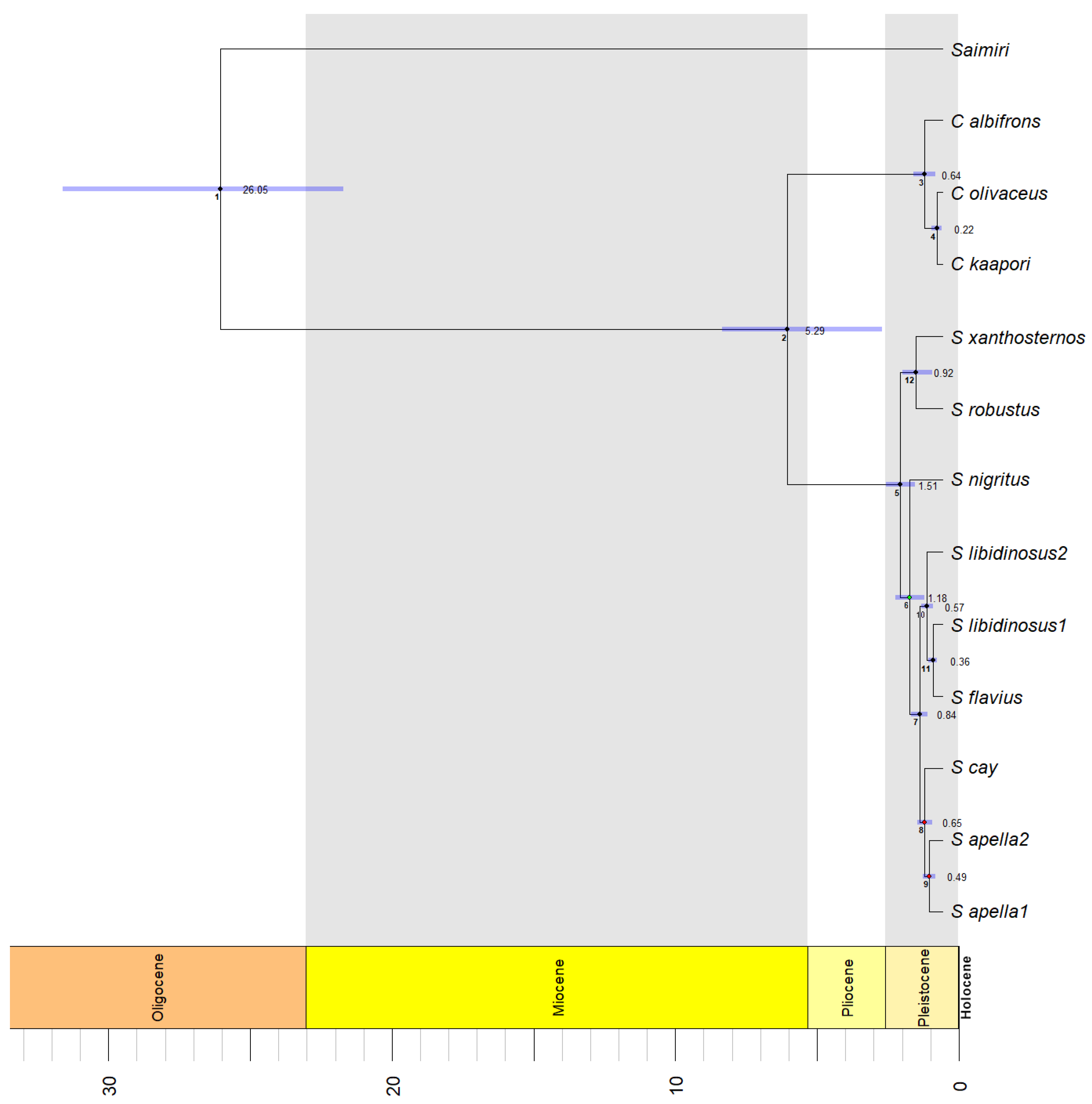
3.3. Species Delimitation and Divergence Dating
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hill, W.C.O. Primates: Comparative Anatomy and Taxonomy. IV. Cebidae, Part A; Interscience Publishers: Geneva, Switzerland, 1960. [Google Scholar]
- Rylands, A.B.; Schneider, H.; Langguth, A.; Mittermeier, R.A.; Groves, C.P.; Rodríguez-Luna, E. An Assessment of the Diversity of New World Primates. Neotrop. Primates 2000, 8, 61–93. [Google Scholar]
- Silva Júnior, J.S. Especiação Nos Macacos-Prego e Caiararas, Gênero Cebus Erxleben, 1777 (Primates, Cebidae); Universidade Federal do Rio de Janeiro: Rio de Janeiro, Brazil, 2001. [Google Scholar]
- Alfaro, J.W.L.; Silva, J.D.S.E.; Rylands, A.B. How Different Are Robust and Gracile Capuchin Monkeys? An Argument for the Use of Sapajus and Cebus: Sapajus and Cebus. Am. J. Primatol. 2012, 74, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Lima, M.G.M.; Buckner, J.C.; Silva-Júnior, J.d.S.e.; Aleixo, A.; Martins, A.B.; Boubli, J.P.; Link, A.; Farias, I.P.; da Silva, M.N.; Röhe, F.; et al. Capuchin Monkey Biogeography: Understanding Sapajus Pleistocene Range Expansion and the Current Sympatry between Cebus and Sapajus. J. Biogeogr. 2017, 44, 810–820. [Google Scholar] [CrossRef]
- Fragaszy, D.M.; Fedigan, L.M.; Visalberghi, E. The Complete Capuchin: The Biology of the Genus Cebus; Cambridge University Press: Cambridge, UK, 2004; ISBN 978-0-521-66768-5. [Google Scholar]
- Rylands, A.B.; Bezerra, B.M.; Paim, F.P.; Queiroz, H. Species Accounts of Cebidae. In Handbook of the Mammals of the World, 3 (Primates); Lynx Edicions: Barcelona, Spain, 2013; pp. 390–413. [Google Scholar]
- Elliot, D.G. A Review of Primates; American Museum of Natural History: New York, NY, USA, 1913. [Google Scholar]
- Hershkovitz, P. Mammals of Northern Colombia, Preliminary Report No. 4: Monkeys (Primates), with Taxonomic Revisions of Some Forms. Proc. United States Natl. Mus. 1949, 98, 323–427, Plates 15–17, Figures 52–59. [Google Scholar] [CrossRef]
- Cabrera, A. Catálogo de Los Mamíferos de América Del Sur I (Metatheria, Unguiculata, Carnívora). In Revista del Museo Argentino de Ciencias Naturales “Bernardino Rivadavia” e Instituto Nacional de Investigación de las Ciencias Naturales; Ciencias zoológicas; Casa Editora Coni: Buenos Aires, Argentina, 1957; Volume 4, pp. 1–307. [Google Scholar]
- Groves, C.P. Primate Taxonomy. In Smithsonian Series in Comparative Evolutionary Biology; Smithsonian Institution Press: Washington, DC, USA, 2001; ISBN 1-56098-872-X. [Google Scholar]
- Groves, C.P. Order Primates. In Mammal Species of the World: A Taxonomic and Geographic Reference; Johns Hopkins University Press: Baltimore, MD, USA, 2005; Volume 1, pp. 111–184. [Google Scholar]
- Lima, M.G.M.; Silva-Júnior, J.d.S.e.; Černý, D.; Buckner, J.C.; Aleixo, A.; Chang, J.; Zheng, J.; Alfaro, M.E.; Martins, A.; Di Fiore, A.; et al. A Phylogenomic Perspective on the Robust Capuchin Monkey (Sapajus) Radiation: First Evidence for Extensive Population Admixture across South America. Mol. Phylogenetics Evol. 2018, 124, 137–150. [Google Scholar] [CrossRef]
- Rylands, A.B.; Mittermeier, R.A.; Silva, J.S. Neotropical Primates: Taxonomy and Recently Described Species and Subspecies: Neotropical Primate Taxonomy. Int. Zoo Yearb. 2012, 46, 11–24. [Google Scholar] [CrossRef]
- Oliveira, M.M.; Langguth, A. Rediscovery of Marcgrave’s Capuchin Monkey and Designation of a Neotype for Simia Flavia Schreber, 1774 (Primates, Cebidae). Bol. Mus. Nac. 2006, 523, 1–16. [Google Scholar]
- Ferreira, R.G.; Jerusalinsky, L.; Silva, T.C.F.; de Souza Fialho, M.; de Araújo Roque, A.; Fernandes, A.; Arruda, F. On the Occurrence of Cebus flavius (Schreber 1774) in the Caatinga, and the Use of Semi-Arid Environments by Cebus Species in the Brazilian State of Rio Grande Do Norte. Primates 2009, 50, 357–362. [Google Scholar] [CrossRef]
- Fialho, M.d.S.; Valença-Montenegro, M.M.; da Silva, T.C.F.; Ferreira, J.G.; Laroque, P.d.O. Ocorrência de Sapajus Flavius e Alouatta Belzebul No Centro de Endemismo Pernambuco. Neotrop. Primates 2014, 21, 214–218. [Google Scholar] [CrossRef]
- Giarla, T.C.; Esselstyn, J.A. The Challenges of Resolving a Rapid, Recent Radiation: Empirical and Simulated Phylogenomics of Philippine Shrews. Syst. Biol. 2015, 64, 727–740. [Google Scholar] [CrossRef]
- Harris, R.B.; Alström, P.; Ödeen, A.; Leaché, A.D. Discordance between Genomic Divergence and Phenotypic Variation in a Rapidly Evolving Avian Genus (Motacilla). Mol. Phylogenetics Evol. 2018, 120, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Pollock, D.D.; Zwickl, D.J.; McGuire, J.A.; Hillis, D.M. Increased Taxon Sampling Is Advantageous for Phylogenetic Inference. Syst. Biol. 2002, 51, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Zwickl, D.J.; Hillis, D.M. Increased Taxon Sampling Greatly Reduces Phylogenetic Error. Syst. Biol. 2002, 51, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Health, T.A.; Hedtke, S.M.; Hillis, D.M. Taxon Sampling and the Accuracy of Phylogenetic Analyses. J. Syst. Evol. 2008, 46, 239–257. [Google Scholar]
- Kiesling, J.; Yi, S.V.; Xu, K.; Gianluca Sperone, F.; Wildman, D.E. The Tempo and Mode of New World Monkey Evolution and Biogeography in the Context of Phylogenomic Analysis. Mol. Phylogenetics Evol. 2015, 82, 386–399. [Google Scholar] [CrossRef]
- Moyle, R.G.; Filardi, C.E.; Smith, C.E.; Diamond, J. Explosive Pleistocene Diversification and Hemispheric Expansion of a “Great Speciator”. Proc. Natl. Acad. Sci. USA 2009, 106, 1863–1868. [Google Scholar] [CrossRef] [PubMed]
- Nee, S.; Mooers, A.O.; Harvey, P.H. Tempo and Mode of Evolution Revealed from Molecular Phylogenies. Proc. Natl. Acad. Sci. USA 1992, 89, 8322–8326. [Google Scholar] [CrossRef] [PubMed]
- Rundell, R.J.; Price, T.D. Adaptive Radiation, Nonadaptive Radiation, Ecological Speciation and Nonecological Speciation. Trends Ecol. Evol. 2009, 24, 394–399. [Google Scholar] [CrossRef]
- Braun, E.L.; Kimball, R.T. Polytomies, the Power of Phylogenetic Inference, and the Stochastic Nature of Molecular Evolution: A Comment on Walsh et al. (1999). Evolution 2001, 55, 1261. [Google Scholar] [CrossRef]
- Degnan, J.H.; Rosenberg, N.A. Discordance of Species Trees with Their Most Likely Gene Trees. PLoS Genet. 2006, 2, e68. [Google Scholar] [CrossRef]
- Maddison, W.P.; Knowles, L.L. Inferring Phylogeny Despite Incomplete Lineage Sorting. Syst. Biol. 2006, 55, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Knowles, L.L.; Carstens, B.C. Delimiting Species without Monophyletic Gene Trees. Syst. Biol. 2007, 56, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.G.; Larson, E.L. Hybridization, Introgression, and the Nature of Species Boundaries. J. Hered. 2014, 105, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Eckert, A.; Carstens, B. Does Gene Flow Destroy Phylogenetic Signal? The Performance of Three Methods for Estimating Species Phylogenies in the Presence of Gene Flow. Mol. Phylogenetics Evol. 2008, 49, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Fontenot, B.E.; Makowsky, R.; Chippindale, P.T. Nuclear–Mitochondrial Discordance and Gene Flow in a Recent Radiation of Toads. Mol. Phylogenetics Evol. 2011, 59, 66–80. [Google Scholar] [CrossRef]
- Kutschera, V.E.; Bidon, T.; Hailer, F.; Rodi, J.L.; Fain, S.R.; Janke, A. Bears in a Forest of Gene Trees: Phylogenetic Inference Is Complicated by Incomplete Lineage Sorting and Gene Flow. Mol. Biol. Evol. 2014, 31, 2004–2017. [Google Scholar] [CrossRef] [PubMed]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-Wide Genetic Marker Discovery and Genotyping Using next-Generation Sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef]
- McCormack, J.E.; Hird, S.M.; Zellmer, A.J.; Carstens, B.C.; Brumfield, R.T. Applications of Next-Generation Sequencing to Phylogeography and Phylogenetics. Mol. Phylogenetics Evol. 2013, 66, 526–538. [Google Scholar] [CrossRef]
- Ekblom, R.; Galindo, J. Applications of next Generation Sequencing in Molecular Ecology of Non-Model Organisms. Heredity 2011, 107, 1–15. [Google Scholar] [CrossRef]
- Van Orsouw, N.J.; Hogers, R.C.J.; Janssen, A.; Yalcin, F.; Snoeijers, S.; Verstege, E.; Schneiders, H.; van der Poel, H.; van Oeveren, J.; Verstegen, H.; et al. Complexity Reduction of Polymorphic Sequences (CRoPSTM): A Novel Approach for Large-Scale Polymorphism Discovery in Complex Genomes. PLoS ONE 2007, 2, e1172. [Google Scholar] [CrossRef]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Van Tassell, C.P.; Smith, T.P.L.; Matukumalli, L.K.; Taylor, J.F.; Schnabel, R.D.; Lawley, C.T.; Haudenschild, C.D.; Moore, S.S.; Warren, W.C.; Sonstegard, T.S. SNP Discovery and Allele Frequency Estimation by Deep Sequencing of Reduced Representation Libraries. Nat. Methods 2008, 5, 247–252. [Google Scholar] [CrossRef]
- Davey, J.W.; Blaxter, M.L. RADSeq: Next-Generation Population Genetics. Brief. Funct. Genom. 2010, 9, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Bergey, C.M.; Pozzi, L.; Disotell, T.R.; Burrell, A.S. A New Method for Genome-Wide Marker Development and Genotyping Holds Great Promise for Molecular Primatology. Int. J. Primatol. 2013, 34, 303–314. [Google Scholar] [CrossRef]
- Reitzel, A.M.; Herrera, S.; Layden, M.J.; Martindale, M.Q.; Shank, T.M. Going Where Traditional Markers Have Not Gone before: Utility of and Promise for RAD Sequencing in Marine Invertebrate Phylogeography and Population Genomics. Mol. Ecol. 2013, 22, 2953–2970. [Google Scholar] [CrossRef]
- Hohenlohe, P.A.; Day, M.D.; Amish, S.J.; Miller, M.R.; Kamps-Hughes, N.; Boyer, M.C.; Muhlfeld, C.C.; Allendorf, F.W.; Johnson, E.A.; Luikart, G. Genomic Patterns of Introgression in Rainbow and Westslope Cutthroat Trout Illuminated by Overlapping Paired-End RAD Sequencing. Mol. Ecol. 2013, 22, 3002–3013. [Google Scholar] [CrossRef]
- Jones, J.C.; Fan, S.; Franchini, P.; Schartl, M.; Meyer, A. The Evolutionary History of Xiphophorus Fish and Their Sexually Selected Sword: A Genome-Wide Approach Using Restriction Site-Associated DNA Sequencing. Mol. Ecol. 2013, 22, 2986–3001. [Google Scholar] [CrossRef]
- Takahashi, T.; Nagano, A.J.; Kawaguchi, L.; Onikura, N.; Nakajima, J.; Miyake, T.; Suzuki, N.; Kanoh, Y.; Tsuruta, T.; Tanimoto, T.; et al. A DdRAD-Based Population Genetics and Phylogenetics of an Endangered Freshwater Fish from Japan. Conserv. Genet. 2020, 21, 641–652. [Google Scholar] [CrossRef]
- Nadeau, N.J.; Ruiz, M.; Salazar, P.; Counterman, B.; Medina, J.A.; Ortiz-Zuazaga, H.; Morrison, A.; McMillan, W.O.; Jiggins, C.D.; Papa, R. Population Genomics of Parallel Hybrid Zones in the Mimetic Butterflies, H. Melpomene and H. Erato. Genome Res. 2014, 24, 1316–1333. [Google Scholar] [CrossRef]
- Kozlov, M.V.; Mutanen, M.; Lee, K.M.; Huemer, P. Cryptic Diversity in the Long-Horn Moth Nemophora degeerella (Lepidoptera: Adelidae) Revealed by Morphology, DNA Barcodes and Genome-Wide DdRAD-Seq Data: Cryptic Diversity in Nemophora degeerella. Syst. Entomol. 2017, 42, 329–346. [Google Scholar] [CrossRef]
- Lavretsky, P.; DaCosta, J.M.; Sorenson, M.D.; McCracken, K.G.; Peters, J.L. DdRAD-seq Data Reveal Significant Genome-wide Population Structure and Divergent Genomic Regions That Distinguish the Mallard and Close Relatives in North America. Mol. Ecol. 2019, 28, 2594–2609. [Google Scholar] [CrossRef] [PubMed]
- Lah, L.; Trense, D.; Benke, H.; Berggren, P.; Gunnlaugsson, P.; Lockyer, C.; Öztürk, A.; Öztürk, B.; Pawliczka, I.; Roos, A.; et al. Spatially Explicit Analysis of Genome-Wide SNPs Detects Subtle Population Structure in a Mobile Marine Mammal, the Harbor Porpoise. PLoS ONE 2016, 11, e0162792. [Google Scholar] [CrossRef]
- Mynhardt, S.; Bennett, N.C.; Bloomer, P. New Insights from RADseq Data on Differentiation in the Hottentot Golden Mole Species Complex from South Africa. Mol. Phylogenetics Evol. 2020, 143, 106667. [Google Scholar] [CrossRef] [PubMed]
- Scally, A.; Yngvadottir, B.; Xue, Y.; Ayub, Q.; Durbin, R.; Tyler-Smith, C. A Genome-Wide Survey of Genetic Variation in Gorillas Using Reduced Representation Sequencing. PLoS ONE 2013, 8, e65066. [Google Scholar] [CrossRef]
- Ennes Silva, F.; Valsecchi do Amaral, J.; Roos, C.; Bowler, M.; Röhe, F.; Sampaio, R.; Cora Janiak, M.; Bertuol, F.; Ismar Santana, M.; de Souza Silva Júnior, J.; et al. Molecular Phylogeny and Systematics of Bald Uakaris, Genus Cacajao (Primates: Pitheciidae), with the Description of a New Species. Mol. Phylogenetics Evol. 2022, 173, 107509. [Google Scholar] [CrossRef]
- Costa-Araújo, R.; de Melo, F.R.; Canale, G.R.; Hernández-Rangel, S.M.; Messias, M.R.; Rossi, R.V.; Silva, F.E.; da Silva, M.N.F.; Nash, S.D.; Boubli, J.P.; et al. The Munduruku Marmoset: A New Monkey Species from Southern Amazonia. PeerJ 2019, 7, e7019. [Google Scholar] [CrossRef]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non-Model Species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef]
- Buckner, J.C.; Jack, K.M.; Melin, A.D.; Schoof, V.A.M.; Gutiérrez-Espeleta, G.A.; Lima, M.G.M.; Lynch, J.W. Major Histocompatibility Complex Class II DR and DQ Evolution and Variation in Wild Capuchin Monkey Species (Cebinae). PLoS ONE 2021, 16, e0254604. [Google Scholar] [CrossRef]
- Valencia, L.M.; Martins, A.; Ortiz, E.M.; Di Fiore, A. A RAD-Sequencing Approach to Genome-Wide Marker Discovery, Genotyping, and Phylogenetic Inference in a Diverse Radiation of Primates. PLoS ONE 2018, 13, e0201254. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 10 April 2023).
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner. In Proceedings of the 9th Annual Genomics of Energy & Environment Meeting, Walnut Creek, CA, USA, 17–20 March 2014. [Google Scholar]
- Renaud, G.; Stenzel, U.; Maricic, T.; Wiebe, V.; Kelso, J. DeML: Robust Demultiplexing of Illumina Sequences Using a Likelihood-Based Approach. Bioinformatics 2015, 31, 770–772. [Google Scholar] [CrossRef] [PubMed]
- Eaton, D.A.R.; Overcast, I. Ipyrad: Interactive Assembly and Analysis of RADseq Datasets. Bioinformatics 2020, 36, 2592–2594. [Google Scholar] [CrossRef] [PubMed]
- Byrne, H.; Webster, T.H.; Brosnan, S.F.; Izar, P.; Lynch, J.W. Signatures of Adaptive Evolution in Platyrrhine Primate Genomes. Proc. Natl. Acad. Sci. USA 2022, 119, e2116681119. [Google Scholar] [CrossRef]
- Fumagalli, M. Assessing the Effect of Sequencing Depth and Sample Size in Population Genetics Inferences. PLoS ONE 2013, 8, e79667. [Google Scholar] [CrossRef] [PubMed]
- Harvey, M.G.; Judy, C.D.; Seeholzer, G.F.; Maley, J.M.; Graves, G.R.; Brumfield, R.T. Similarity Thresholds Used in DNA Sequence Assembly from Short Reads Can Reduce the Comparability of Population Histories across Species. PeerJ 2015, 3, e895. [Google Scholar] [CrossRef]
- Rubin, B.E.R.; Ree, R.H.; Moreau, C.S. Inferring Phylogenies from RAD Sequence Data. PLoS ONE 2012, 7, e33394. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- De Queiroz, A.; Gatesy, J. The Supermatrix Approach to Systematics. Trends Ecol. Evol. 2007, 22, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yu, L.; Pearl, D.K.; Edwards, S.V. Estimating Species Phylogenies Using Coalescence Times among Sequences. Syst. Biol. 2009, 58, 468–477. [Google Scholar] [CrossRef]
- Liu, L.; Wu, S.; Yu, L. Coalescent Methods for Estimating Species Trees from Phylogenomic Data: Estimating Species Trees from Phylogenomic Data. J. Syst. Evol. 2015, 53, 380–390. [Google Scholar] [CrossRef]
- Bryant, D.; Bouckaert, R.; Felsenstein, J.; Rosenberg, N.A.; RoyChoudhury, A. Inferring Species Trees Directly from Biallelic Genetic Markers: Bypassing Gene Trees in a Full Coalescent Analysis. Mol. Biol. Evol. 2012, 29, 1917–1932. [Google Scholar] [CrossRef] [PubMed]
- Chifman, J.; Kubatko, L. Quartet Inference from SNP Data Under the Coalescent Model. Bioinformatics 2014, 30, 3317–3324. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Carstens, B.C.; Pelletier, T.A.; Reid, N.M.; Satler, J.D. How to Fail at Species Delimitation. Mol. Ecol. 2013, 22, 4369–4383. [Google Scholar] [CrossRef] [PubMed]
- Leache, A.D.; Fujita, M.K.; Minin, V.N.; Bouckaert, R.R. Species Delimitation Using Genome-Wide SNP Data. Syst. Biol. 2014, 63, 534–542. [Google Scholar] [CrossRef]
- Fujita, M.K.; Leaché, A.D.; Burbrink, F.T.; McGuire, J.A.; Moritz, C. Coalescent-Based Species Delimitation in an Integrative Taxonomy. Trends Ecol. Evol. 2012, 27, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Grummer, J.A.; Bryson, R.W.; Reeder, T.W. Species Delimitation Using Bayes Factors: Simulations and Application to the Sceloporus scalaris Species Group (Squamata: Phrynosomatidae). Syst. Biol. 2014, 63, 119–133. [Google Scholar] [CrossRef]
- Edwards, S.V.; Xi, Z.; Janke, A.; Faircloth, B.C.; McCormack, J.E.; Glenn, T.C.; Zhong, B.; Wu, S.; Lemmon, E.M.; Lemmon, A.R.; et al. Implementing and Testing the Multispecies Coalescent Model: A Valuable Paradigm for Phylogenomics. Mol. Phylogenetics Evol. 2016, 94, 447–462. [Google Scholar] [CrossRef]
- Afonso Silva, A.C.; Santos, N.; Ogilvie, H.A.; Moritz, C. Validation and Description of Two New North-Western Australian Rainbow Skinks with Multispecies Coalescent Methods and Morphology. PeerJ 2017, 5, e3724. [Google Scholar] [CrossRef] [PubMed]
- Solano-Zavaleta, I.; Nieto-Montes de Oca, A. Species Limits in the Morelet’s Alligator Lizard (Anguidae: Gerrhonotinae). Mol. Phylogenetics Evol. 2018, 120, 16–27. [Google Scholar] [CrossRef]
- Kass, R.E.; Raftery, A.E. Bayes Factors. J. Am. Stat. Assoc. 1995, 90, 773–795. [Google Scholar] [CrossRef]
- Oaks, J.R.; Cobb, K.A.; Minin, V.N.; Leaché, A.D. Marginal Likelihoods in Phylogenetics: A Review of Methods and Applications. Syst. Biol. 2019, 68, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, H.A.; Bouckaert, R.R.; Drummond, A.J. StarBEAST2 Brings Faster Species Tree Inference and Accurate Estimates of Substitution Rates. Mol. Biol. Evol. 2017, 34, 2101–2114. [Google Scholar] [CrossRef]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N.; et al. BEAST 2.5: An Advanced Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [PubMed]
- Aydin, Z.; Marcussen, T.; Ertekin, A.S.; Oxelman, B. Marginal Likelihood Estimate Comparisons to Obtain Optimal Species Delimitations in Silene Sect. Cryptoneurae (Caryophyllaceae). PLoS ONE 2014, 9, e106990. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for Inference of Large Phylogenetic Trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Kay, R.F. Biogeography in Deep Time—What Do Phylogenetics, Geology, and Paleoclimate Tell Us about Early Platyrrhine Evolution? Mol. Phylogenetics Evol. 2015, 82, 358–374. [Google Scholar] [CrossRef]
- Bloch, J.I.; Woodruff, E.D.; Wood, A.R.; Rincon, A.F.; Harrington, A.R.; Morgan, G.S.; Foster, D.A.; Montes, C.; Jaramillo, C.A.; Jud, N.A.; et al. First North American Fossil Monkey and Early Miocene Tropical Biotic Interchange. Nature 2016, 533, 243–246. [Google Scholar] [CrossRef]
- Di Fiore, A.; Chaves, P.B.; Cornejo, F.M.; Schmitt, C.A.; Shanee, S.; Cortés-Ortiz, L.; Fagundes, V.; Roos, C.; Pacheco, V. The Rise and Fall of a Genus: Complete MtDNA Genomes Shed Light on the Phylogenetic Position of Yellow-Tailed Woolly Monkeys, Lagothrix flavicauda, and on the Evolutionary History of the Family Atelidae (Primates: Platyrrhini). Mol. Phylogenetics Evol. 2015, 82, 495–510. [Google Scholar] [CrossRef]
- Silva, T.C.F. Estudo Da Variação Na Pelagem e Da Distribuição Geográfica Em Cebus flavius (Schreber, 1774) e Cebus libidinosus (Spix, 1823) Do Nordeste Do Brasil. Master Thesis, Universidade Federal da Paraíba, João Pessoa, Brazil, 2010. [Google Scholar]
- Ruiz-García, M.; Castillo, M.I.; Luengas-Villamil, K. It Is Misleading to Use Sapajus (Robust Capuchins) as a Genus? A Review of the Evolution of the Capuchins and Suggestions on Their Systematics. In Phylogeny, Molecular Population Genetics, Evolutionary Biology and Conservation of the Neotropical Primates; Nova Science Publisher Inc.: New York, NY, USA, 2016; pp. 209–268. [Google Scholar]
- Ruiz-García, M.; Castillo, M.I.; Lichilín-Ortiz, N.; Pinedo-Castro, M. Molecular Relationships and Classification of Several Tufted Capuchin Lineages (Cebus apella, Cebus xanthosternos and Cebus nigritus, Cebidae), by Means of Mitochondrial Cytochrome Oxidase II Gene Sequences. IJFP 2012, 83, 100–125. [Google Scholar] [CrossRef] [PubMed]
- Lynch Alfaro, J.W.; Boubli, J.P.; Olson, L.E.; Di Fiore, A.; Wilson, B.; Gutiérrez-Espeleta, G.A.; Chiou, K.L.; Schulte, M.; Neitzel, S.; Ross, V.; et al. Explosive Pleistocene Range Expansion Leads to Widespread Amazonian Sympatry between Robust and Gracile Capuchin Monkeys: Biogeography of Neotropical Capuchin Monkeys. J. Biogeogr. 2012, 39, 272–288. [Google Scholar] [CrossRef]
- Boubli, J.P.; Ribas, C.; Lynch Alfaro, J.W.; Alfaro, M.E.; da Silva, M.N.F.; Pinho, G.M.; Farias, I.P. Spatial and Temporal Patterns of Diversification on the Amazon: A Test of the Riverine Hypothesis for All Diurnal Primates of Rio Negro and Rio Branco in Brazil. Mol. Phylogenetics Evol. 2015, 82, 400–412. [Google Scholar] [CrossRef] [PubMed]
- Lynch Alfaro, J.W.; Boubli, J.P.; Paim, F.P.; Ribas, C.C.; da Silva, M.N.F.; Messias, M.R.; Röhe, F.; Mercês, M.P.; Silva Júnior, J.S.; Silva, C.R.; et al. Biogeography of Squirrel Monkeys (Genus saimiri): South-Central Amazon Origin and Rapid Pan-Amazonian Diversification of a Lowland Primate. Mol. Phylogenetics Evol. 2015, 82, 436–454. [Google Scholar] [CrossRef] [PubMed]
- Piel, A.K.; Stewart, F.A.; Pintea, L.; Li, Y.; Ramirez, M.A.; Loy, D.E.; Crystal, P.A.; Learn, G.H.; Knapp, L.A.; Sharp, P.M.; et al. The Malagarasi River Does Not Form an Absolute Barrier to Chimpanzee Movement in Western Tanzania. PLoS ONE 2013, 8, e58965. [Google Scholar] [CrossRef] [PubMed]
- Link, A.; Valencia, L.M.; Céspedes, L.N.; Duque, L.D.; Cadena, C.D.; Di Fiore, A. Phylogeography of the Critically Endangered Brown Spider Monkey (Ateles hybridus): Testing the Riverine Barrier Hypothesis. Int. J. Primatol. 2015, 36, 530–547. [Google Scholar] [CrossRef]
- Leaché, A.D.; Harris, R.B.; Rannala, B.; Yang, Z. The Influence of Gene Flow on Species Tree Estimation: A Simulation Study. Syst. Biol. 2014, 63, 17–30. [Google Scholar] [CrossRef]
- Tajima, F. Evolutionary Relationship of DNA Sequences in Finite Populations. Genetics 1983, 105, 437–460. [Google Scholar] [CrossRef]
- Pamilo, P.; Nei, M. Relationships between Gene Trees and Species Trees. Mol. Biol. Evol. 1988, 5, 568–583. [Google Scholar] [CrossRef]
- Heled, J.; Drummond, A.J. Bayesian Inference of Species Trees from Multilocus Data. Mol. Biol. Evol. 2010, 27, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Martins-Junior, A.M.G.; Amorim, N.; Carneiro, J.C.; de Mello Affonso, P.R.A.; Sampaio, I.; Schneider, H. Alu Elements and the Phylogeny of Capuchin (Cebus and Sapajus) Monkeys: Alu Elements and Capuchin Monkeys. Am. J. Primatol. 2015, 77, 368–375. [Google Scholar] [CrossRef] [PubMed]
- De Queiroz, K. Species Concepts and Species Delimitation. Syst. Biol. 2007, 56, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.L.; Knowles, L.L. Species Detection and Individual Assignment in Species Delimitation: Can Integrative Data Increase Efficacy? Proc. R. Soc. B 2014, 281, 20132765. [Google Scholar] [CrossRef]
- Perelman, P.; Johnson, W.E.; Roos, C.; Seuánez, H.N.; Horvath, J.E.; Moreira, M.A.M.; Kessing, B.; Pontius, J.; Roelke, M.; Rumpler, Y.; et al. A Molecular Phylogeny of Living Primates. PLoS Genet. 2011, 7, e1001342. [Google Scholar] [CrossRef]
- Springer, M.S.; Meredith, R.W.; Gatesy, J.; Emerling, C.A.; Park, J.; Rabosky, D.L.; Stadler, T.; Steiner, C.; Ryder, O.A.; Janečka, J.E.; et al. Macroevolutionary Dynamics and Historical Biogeography of Primate Diversification Inferred from a Species Supermatrix. PLoS ONE 2012, 7, e49521. [Google Scholar] [CrossRef] [PubMed]
- Lynch Alfaro, J.W.; Cortés-Ortiz, L.; Di Fiore, A.; Boubli, J.P. Special Issue: Comparative Biogeography of Neotropical Primates. Mol. Phylogenetics Evol. 2015, 82, 518–529. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Minimum % to Call a Locus 1 | Minimum # to Call a Locus 2 | Number of Loci 3 | SNPs Matrix Size | % Missing SNPs | Total # of Variable Sites | Parsimony- Informative Sites 4 | % Missing Sequence Matrix |
|---|---|---|---|---|---|---|---|
| ≈90% | 150 | 16,880 | 353,995 | 15.38% | 327,622 | 178,012 | 20.4% |
| ≈75% | 130 | 31,216 | 614,050 | 19.34% | 572,259 | 299,054 | 24.2% |
| ≈60% | 100 | 43,736 | 830,442 | 24.41% | 777,155 | 394,844 | 29.2% |
| ≈30% | 50 | 64,081 | 1,125,828 | 34.32% | 1,054,796 | 518,460 | 40.1% |
| Model Hypothesis 1 (Ranked by MLE) | Marginal Likelihood Estimate (MLE) | Bayes Factor (2lnBF) 2 to the 1st Ranked Model H7 | Bayes Factor (2lnBF) 2 to the 2nd Ranked Model H5 |
|---|---|---|---|
| H7 | −185,731.7644 | - | −192.38 |
| H5 | −185,827.9526 | 192.38 | - |
| H6 | −186,006.9226 | 550.32 | 357.94 |
| H1 | −186,153.4149 | 843.30 | 650.92 |
| H4 | −186,337.5626 | 1211.60 | 1019.22 |
| H3 | −186,424.9737 | 1386.42 | 1194.04 |
| H1 | −187,530.4374 | 3597.35 | 3404.97 |
| H0 | −189,165.7362 | 6867.94 | 6675.57 |
| Node in Time Tree 1 | Model Calibration 1 | Model Calibration 2 | Model Calibration 3 2 | |||
|---|---|---|---|---|---|---|
| Median | 95% HPD | Median | 95% HPD | Median | 95% HPD | |
| 1 | 13.04 | 12.1–16.39 | 14.03 | 13.08–15.46 | 26.05 | 21.71–31.62 |
| 2 | 2.85 | 0.9–3.89 | 3.08 | 1.26–3.83 | 5.29 | 1.95–7.58 |
| 3 | 0.34 | 0.13–0.53 | 0.37 | 0.16–0.53 | 0.64 | 0.26–1.03 |
| 4 | 0.11 | 0.02–0.2 | 0.12 | 0.02–0.21 | 0.22 | 0.04–0.41 |
| 5 | 0.78 | 0.52–1.07 | 0.83 | 0.57–1.04 | 1.51 | 0.99–2.02 |
| 6 | 0.61 | 0.34–0.88 | 0.65 | 0.4–0.88 | 1.18 | 0.66–1.69 |
| 7 | 0.43 | 0.29–0.59 | 0.46 | 0.33–0.58 | 0.84 | 0.56–1.14 |
| 8 | 0.33 | 0.21–0.47 | 0.35 | 0.23–0.48 | 0.65 | 0.39–0.9 |
| 9 | 0.25 | 0.13–0.37 | 0.26 | 0.16–0.38 | 0.49 | 0.28–0.72 |
| 10 | 0.29 | 0.18–0.4 | 0.32 | 0.21–0.39 | 0.57 | 0.35–0.75 |
| 11 | 0.19 | 0.1–0.27 | 0.2 | 0.13–0.26 | 0.36 | 0.21–0.51 |
| 12 | 0.5 | 0.17–0.74 | 0.53 | 0.2–0.72 | 0.92 | 0.36–1.42 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, A.B.; Valença-Montenegro, M.M.; Lima, M.G.M.; Lynch, J.W.; Svoboda, W.K.; Silva-Júnior, J.d.S.e.; Röhe, F.; Boubli, J.P.; Fiore, A.D. A New Assessment of Robust Capuchin Monkey (Sapajus) Evolutionary History Using Genome-Wide SNP Marker Data and a Bayesian Approach to Species Delimitation. Genes 2023, 14, 970. https://doi.org/10.3390/genes14050970
Martins AB, Valença-Montenegro MM, Lima MGM, Lynch JW, Svoboda WK, Silva-Júnior JdSe, Röhe F, Boubli JP, Fiore AD. A New Assessment of Robust Capuchin Monkey (Sapajus) Evolutionary History Using Genome-Wide SNP Marker Data and a Bayesian Approach to Species Delimitation. Genes. 2023; 14(5):970. https://doi.org/10.3390/genes14050970
Chicago/Turabian StyleMartins, Amely Branquinho, Mônica Mafra Valença-Montenegro, Marcela Guimarães Moreira Lima, Jessica W. Lynch, Walfrido Kühl Svoboda, José de Sousa e Silva-Júnior, Fábio Röhe, Jean Philippe Boubli, and Anthony Di Fiore. 2023. "A New Assessment of Robust Capuchin Monkey (Sapajus) Evolutionary History Using Genome-Wide SNP Marker Data and a Bayesian Approach to Species Delimitation" Genes 14, no. 5: 970. https://doi.org/10.3390/genes14050970
APA StyleMartins, A. B., Valença-Montenegro, M. M., Lima, M. G. M., Lynch, J. W., Svoboda, W. K., Silva-Júnior, J. d. S. e., Röhe, F., Boubli, J. P., & Fiore, A. D. (2023). A New Assessment of Robust Capuchin Monkey (Sapajus) Evolutionary History Using Genome-Wide SNP Marker Data and a Bayesian Approach to Species Delimitation. Genes, 14(5), 970. https://doi.org/10.3390/genes14050970