
Comparative Benchmarking of Optical Genome Mapping and Chromosomal Microarray Reveals High Technological Concordance in CNV Identification and Additional Structural Variant Refinement

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cohort Composition
2.2. Optical Genome Mapping
2.3. Data Analysis
3. Results
3.1. Concordance
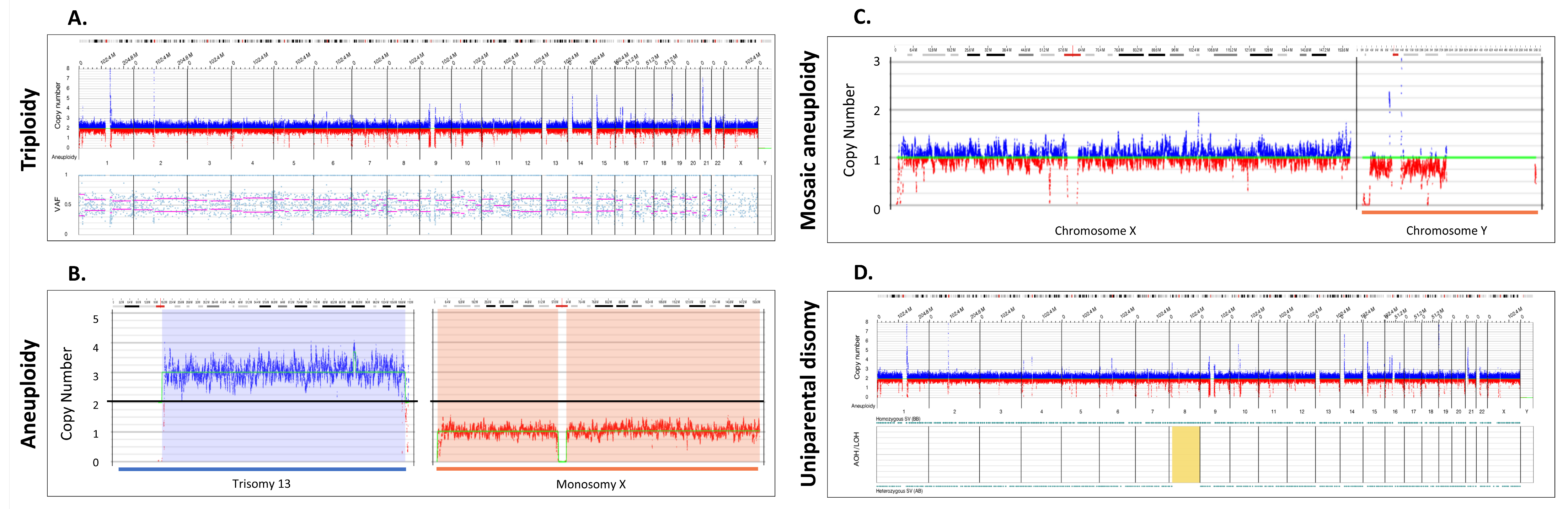
3.2. Detection of Whole Chromosome Copy Gains and Losses and Copy Neutral Events
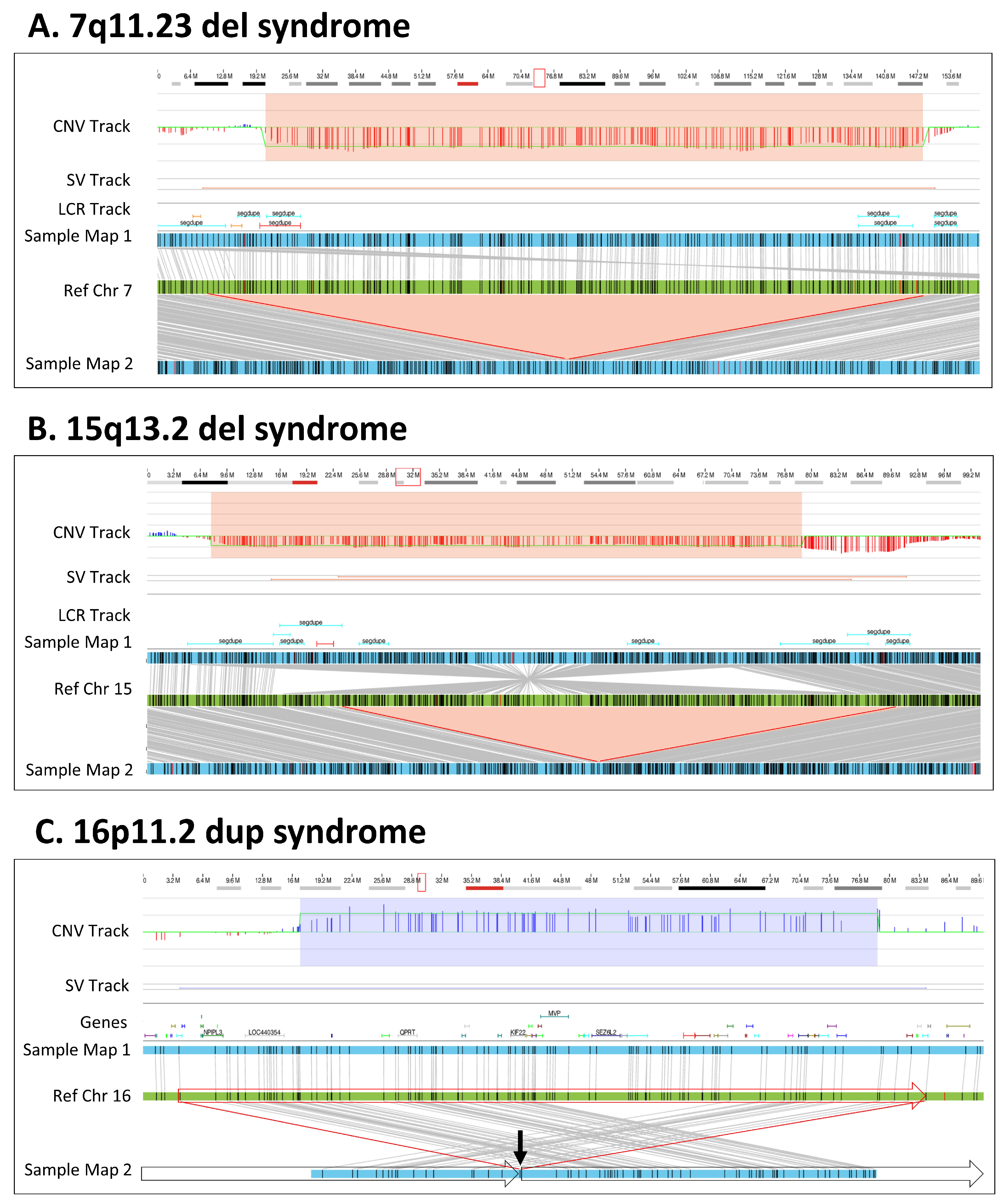
3.3. Microdeletion/Duplication Syndromes
3.4. OGM Resolves the Genomic Structure of CNVs
3.5. OGM Filtration Criteria Used to Select for Potential Pathogenic SVs
4. Discussion
4.1. Strengths and Limitations of OGM
4.2. CNV Size Discrepancies between CMA and OGM
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD). 2020. Available online: https://omim.org/ (accessed on 1 August 2022).
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Manning, M.; Hudgins, L. Array-Based Technology and Recommendations for Utilization in Medical Genetics Practice for Detection of Chromosomal Abnormalities. Genet. Med. 2010, 12, 742–745. [Google Scholar] [CrossRef] [PubMed]
- Shevell, M.; Ashwal, S.; Donley, D.; Flint, J.; Gingold, M.; Hirtz, D.; Majnemer, A.; Noetzel, M.; Sheth, R.D. Practice Parameter: Evaluation of the Child with Global Developmental Delay [RETIRED]: Report of the Quality Standards Subcommittee of the American Academy of Neurology and The Practice Committee of the Child Neurology Society. Neurology 2003, 60, 367–380. [Google Scholar] [CrossRef]
- Moeschler, J.B.; Shevell, M.; Committee on Genetics; Moeschler, J.B.; Shevell, M.; Saul, R.A.; Chen, E.; Freedenberg, D.L.; Hamid, R.; Jones, M.C.; et al. Comprehensive Evaluation of the Child with Intellectual Disability or Global Developmental Delays. Pediatrics 2014, 134, e903–e918. [Google Scholar] [CrossRef]
- Hyman, S.L.; Levy, S.E.; Myers, S.M.; Council on Children with Disabilities, Section on Developmental and Behavioral Pediatrics; Kuo, D.Z.; Apkon, S.; Davidson, L.F.; Ellerbeck, K.A.; Foster, J.E.A.; Noritz, G.H.; et al. Identification, Evaluation, and Management of Children with Autism Spectrum Disorder. Pediatrics 2020, 145, e20193447. [Google Scholar] [CrossRef]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical Standards for the Interpretation and Reporting of Constitutional Copy-Number Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef]
- Waggoner, D.; Wain, K.E.; Dubuc, A.M.; Conlin, L.; Hickey, S.E.; Lamb, A.N.; Martin, C.L.; Morton, C.C.; Rasmussen, K.; Schuette, J.L.; et al. Yield of Additional Genetic Testing after Chromosomal Microarray for Diagnosis of Neurodevelopmental Disability and Congenital Anomalies: A Clinical Practice Resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Bionano Genomics. Bionano Solve Theory of Operation: Structural Variant Calling. 2020. Available online: https://bionanogenomics.com/wp-content/uploads/2018/04/30190-Bionano-Solve-Theory-of-Operation-Variant-Annotation-Pipeline.pdf (accessed on 1 August 2022).
- Chaisson, M.J.P.; Sanders, A.D.; Zhao, X.; Malhotra, A.; Porubsky, D.; Rausch, T.; Gardner, E.J.; Rodriguez, O.L.; Guo, L.; Collins, R.L.; et al. Multi-Platform Discovery of Haplotype-Resolved Structural Variation in Human Genomes. Nat. Commun. 2019, 10, 1784. [Google Scholar] [CrossRef]
- Levy-Sakin, M.; Pastor, S.; Mostovoy, Y.; Li, L.; Leung, A.K.Y.; McCaffrey, J.; Young, E.; Lam, E.T.; Hastie, A.R.; Wong, K.H.Y.; et al. Genome Maps across 26 Human Populations Reveal Population-Specific Patterns of Structural Variation. Nat. Commun. 2019, 10, 1025. [Google Scholar] [CrossRef]
- Bocklandt, S.; Hastie, A.; Cao, H. Bionano Genome Mapping: High-Throughput, Ultra-Long Molecule Genome Analysis System for Precision Genome Assembly and Haploid-Resolved Structural Variation Discovery. In Single Molecule and Single Cell Sequencing; Suzuki, Y., Ed.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; pp. 97–118. ISBN 9789811360374. [Google Scholar]
- Stence, A.A.; Thomason, J.G.; Pruessner, J.A.; Sompallae, R.R.; Snow, A.N.; Ma, D.; Moore, S.A.; Bossler, A.D. Validation of Optical Genome Mapping for the Molecular Diagnosis of Facioscapulohumeral Muscular Dystrophy. J. Mol. Diagn. 2021, 23, 1506–1514. [Google Scholar] [CrossRef]
- Barseghyan, H.; Tang, W.; Wang, R.T.; Almalvez, M.; Segura, E.; Bramble, M.S.; Lipson, A.; Douine, E.D.; Lee, H.; Délot, E.C.; et al. Next-Generation Mapping: A Novel Approach for Detection of Pathogenic Structural Variants with a Potential Utility in Clinical Diagnosis. Genome Med. 2017, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Xu, X.; Ding, L.; Li, H.; Xu, C.; Gong, Y.; Liu, Y.; Mu, T.; Leigh, D.; Cram, D.S.; et al. Clinical Application of Single-Molecule Optical Mapping to a Multigeneration FSHD1 Pedigree. Mol. Genet. Genom. Med. 2019, 7, e565. [Google Scholar] [CrossRef]
- Cope, H.; Barseghyan, H.; Bhattacharya, S.; Fu, Y.; Hoppman, N.; Marcou, C.; Walley, N.; Rehder, C.; Deak, K.; Alkelai, A.; et al. Detection of a Mosaic CDKL5 Deletion and Inversion by Optical Genome Mapping Ends an Exhaustive Diagnostic Odyssey. Mol. Genet. Genom. Med. 2021, 9, e1665. [Google Scholar] [CrossRef]
- Mantere, T.; Neveling, K.; Pebrel-Richard, C.; Benoist, M.; van der Zande, G.; Kater-Baats, E.; Baatout, I.; van Beek, R.; Yammine, T.; Oorsprong, M.; et al. Optical Genome Mapping Enables Constitutional Chromosomal Aberration Detection. Am. J. Hum. Genet. 2021, 108, 1409–1422. [Google Scholar] [CrossRef] [PubMed]
- Mostovoy, Y.; Yilmaz, F.; Chow, S.K.; Chu, C.; Lin, C.; Geiger, E.A.; Meeks, N.J.L.; Chatfield, K.C.; Coughlin, C.R.; Surti, U.; et al. Genomic Regions Associated with Microdeletion/Microduplication Syndromes Exhibit Extreme Diversity of Structural Variation. Genetics 2021, 217, iyaa038. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Xu, J.; Dileep, V.; Zhan, Y.; Song, F.; Le, V.T.; Yardımcı, G.G.; Chakraborty, A.; Bann, D.V.; Wang, Y.; et al. Integrative Detection and Analysis of Structural Variation in Cancer Genomes. Nat. Genet. 2018, 50, 1388–1398. [Google Scholar] [CrossRef]
- Neveling, K.; Mantere, T.; Vermeulen, S.; Oorsprong, M.; van Beek, R.; Kater-Baats, E.; Pauper, M.; van der Zande, G.; Smeets, D.; Weghuis, D.O.; et al. Next-Generation Cytogenetics: Comprehensive Assessment of 52 Hematological Malignancy Genomes by Optical Genome Mapping. Am. J. Hum. Genet. 2021, 108, 1423–1435. [Google Scholar] [CrossRef] [PubMed]
- Kriegova, E.; Fillerova, R.; Minarik, J.; Savara, J.; Manakova, J.; Petrackova, A.; Dihel, M.; Balcarkova, J.; Krhovska, P.; Pika, T.; et al. Whole-Genome Optical Mapping of Bone-Marrow Myeloma Cells Reveals Association of Extramedullary Multiple Myeloma with Chromosome 1 Abnormalities. Sci. Rep. 2021, 11, 14671. [Google Scholar] [CrossRef] [PubMed]
- Kearney, H.M.; Kearney, J.B.; Conlin, L.K. Diagnostic Implications of Excessive Homozygosity Detected by SNP-Based Microarrays: Consanguinity, Uniparental Disomy, and Recessive Single-Gene Mutations. Clin. Lab. Med. 2011, 31, 595–613. [Google Scholar] [CrossRef] [PubMed]
- Lakich, D.; Kazazian, H.H., Jr.; Antonarakis, S.E.; Gitschier, J. Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nat. Genet. 1993, 5, 236–241. [Google Scholar] [CrossRef]
- Coutelier, M.; Holtgrewe, M.; Jäger, M.; Flöttman, R.; Mensah, M.A.; Spielmann, M.; Krawitz, P.; Horn, D.; Beule, D.; Mundlos, S. Combining callers improves the detection of copy number variants from whole-genome sequencing. Eur. J. Hum. Genet. 2022, 30, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Remec, Z.I.; Trebusak Podkrajsek, K.; Repic Lampret, B.; Kovac, J.; Groselj, U.; Tesovnik, T.; Battelino, T.; Debeljak, M. Next-Generation Sequencing in Newborn Screening: A Review of Current State. Front. Genet. 2021, 12, 662254. [Google Scholar] [CrossRef] [PubMed]
- Ewans, L.J.; Minoche, A.E.; Schofield, D.; Shrestha, R.; Puttick, C.; Zhu, Y.; Drew, A.; Gayevskiy, V.; Elakis, G.; Walsh, C.; et al. Whole Exome and Genome Sequencing in Mendelian Disorders: A Diagnostic and Health Economic Analysis. Eur. J. Hum. Genet. 2022, 30, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.A.; Broeckel, U.; Levy, B.; Skinner, S.; Sahajpal, N.S.; Rodriguez, V.; Stence, A.; Awayda, K.; Scharer, G.; Skinner, C.; et al. Multisite Assessment of Optical Genome Mapping for Analysis of Structural Variants in Constitutional Postnatal Cases. J. Mol. Diagn. 2023, 25, 175–188. [Google Scholar] [CrossRef]
- Shieh, J.T.; Penon-Portmann, M.; Wong, K.H.Y.; Levy-Sakin, M.; Verghese, M.; Slavotinek, A.; Gallagher, R.C.; Mendelsohn, B.A.; Tenney, J.; Beleford, D.; et al. Application of Full-Genome Analysis to Diagnose Rare Monogenic Disorders. npj Genom. Med. 2021, 6, 77. [Google Scholar] [CrossRef]
- Smith, A.C.; Neveling, K.; Kanagal-Shamanna, R. Optical genome mapping for structural variation analysis in hematologic malignancies. Am. J. Hematol. 2022, 97, 975–982. [Google Scholar] [CrossRef]
- Levy, B.; Baughn, L.B.; Akkari, Y.; Chartrand, S.; LaBarge, B.; Claxton, D.; Lennon, P.A.; Cujar, C.; Kolhe, R.; Kroeger, K.; et al. Optical genome mapping in acute myeloid leukemia: A multicenter evaluation. Blood Adv. 2023, 7, 1297–1307. [Google Scholar] [CrossRef]
- Sahajpal, N.S.; Mondal, A.K.; Fee, T.; Hilton, B.; Layman, L.; Hastie, A.R.; Chaubey, A.; DuPont, B.R.; Kolhe, R. Clinical Validation and Diagnostic Utility of Optical Genome Mapping in Prenatal Diagnostic Testing. J. Mol. Diagn. 2023, 25, 234–246. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Copy Number Variants | Total | |||
|---|---|---|---|---|---|
| Aneuploidy | Triploidy | Gain | Loss | ||
| CMA | 5 | 2 | 21 | 33 | 61 |
| OGM | 5 | 2 | 20 * | 33 | 60 |
| Concordance | 100% | 100% | 95% | 100% | 98% |
| Sample | CMA | FISH | OGM | Result |
|---|---|---|---|---|
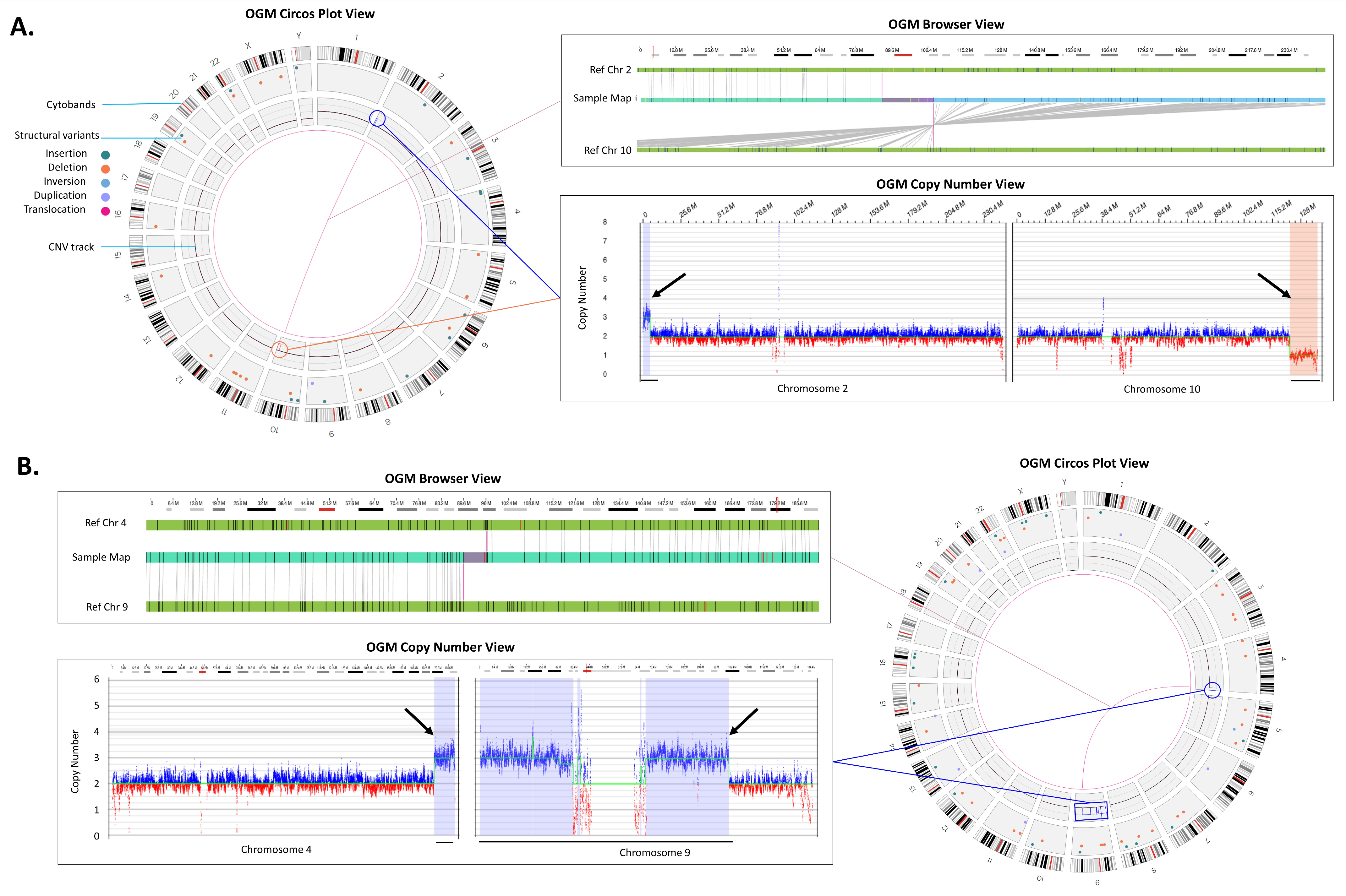
| 36 | arr[GRCh37] 2p25.3p25.2(36400_4801965)x3,10q26.12q26.3(123027554_135403394)x1 | der(10)t(2;10)(p25.2;q26.12) | ogm[GRCh37] t(2;10)(p25.2;q26.1)(4776157;123014483),2p25.3p25.2(15924_4746589)x3,10q26.12q26.3(123011875_135522591)x1 | OGM identified CNVs and translocation. |
| 41 | arr[GRCh37] 4q34.3q35.2(179412576_190896674)x3,9p24.3q31.1(209020_105724992)x3 | +der(9)t(4;9)(q34.3;q31.1) | ogm[GRCh37] t(4;9)(q34.3;q31.1)(179395177;105721182),4q34.3q35.2(179395177_191040751)x3,9p24.3q31.1(14556_105718660)x3, | OGM identified CNVs and translocation. |
| 44 | arr[GRCh37] 1q21.1q21.2(146531538_147726541)x3,9q34.3(139610281_141005513)x1,21q22.13q22.3(38319773_48091215)x3 | der(9)t(9;21)(q34.3;q22.13),(1q21.1)x3 | ogm[GRCh37] t(9;21)(q34.3;q22.13)(136684084;36948511),1q21.1q21.2(146057345_148928812)x3,9q34.3(136472755_138334464)x1,21q22.13q22.3(36948511_46697230)x3 | OGM identified CNVs and translocation. |
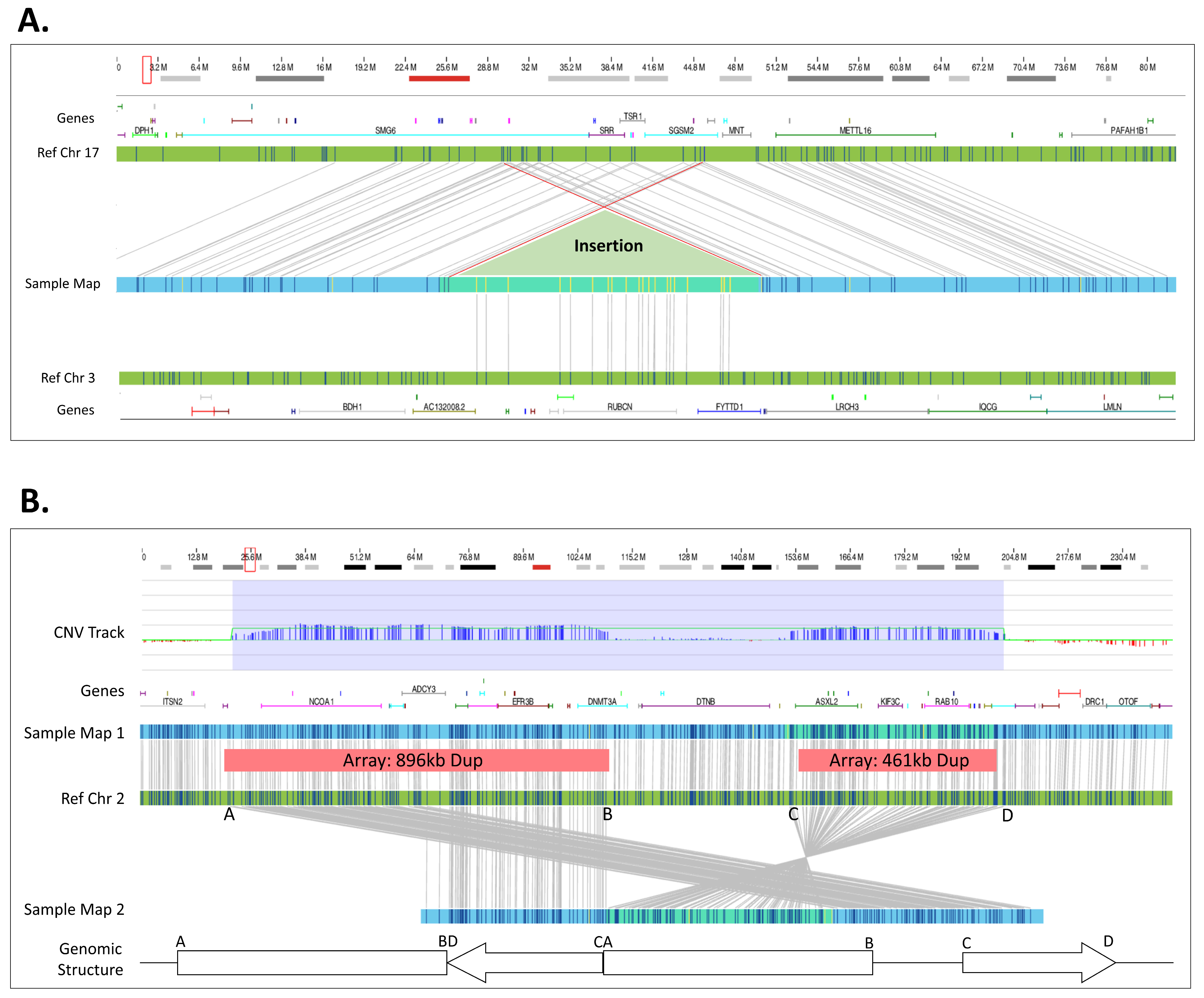
| 26 | arr[GRCh37] 2p11.2(85233220_85575202)x3 | der(2)ins(2)(p?15;p11.2p11.2) | ogm[GRCh37] ins(2;2)(p16.1;p11.2)(57631805;85217155_85572976) | OGM defined CNV insertion site. |
| 28 | arr[GRCh37] 3q29(197398764_197495350)x3 | der(17)ins(17;3)(p13.?3;q29q29) | ogm[GRCh37] ins(17;3)(p13.3;q29)(2155812/2276044;197344236_197495529) | OGM defined CNV insertion site. |
| 30 | arr[GRCh37] 6q14.1(77294196_77479434)x3 | der(X)ins(X;6)(Xq2?8;q14.1q14.1) | ogm[GRCh37] ins(X;6)(q28;q14.1)(152467195;77273466_77477943) | OGM defined CNV insertion site. |
| 31 | arr[GRCh37] 6q14.1(77294196_77479434)x3 | der(X)ins(X;6)(Xq2?8;q14.1q14.1) | ogm[GRCh37] ins(X;6)(q28;q14.1)(152467195;77273466_77477943) | OGM defined CNV insertion site. |
| 24 | arr[GRCh37] 1p36.22(11517159_11892978)x3 | (1p36.22)x3 | ogm[GRCh37] 1p36.22(11518058_11896987)x3 | Tandem duplication (defining orientation) |
| 38 | arr[GRCh37] 16p11.2(29657192_30192346)x3 | (16p11.2)x3 | ogm[GRCh37] 16p11.2(29463027_30202372)x3 | Tandem duplication (defining orientation) |
| 35 | arr[GRCh37] Xp21.3p21.2(28916857_29457146)x2 | (Xp21.3)x3 | ogm[GRCh37] Xp21.3(28912348_29459420)x2 | Tandem duplication (defining orientation) |
| 28 | arr[GRCh37] 6q12(76287632_77298392)x3 | (6q14.1)x3 | ogm[GRCh37] 6q12(76222934_77313055)x3 | Tandem duplication (defining orientation) |
| 40 | arr[GRCh37] 2p23.3(24633371_25529639)x3,2p23.3(25961533_26422725)x3 | (2p23.3)x3 | ogm[GRCh37] 2p23.3(24643804_25517100)x3,2q23.3(25943863_26441430)x3 | OGM identified both CNVs and characterized their structure. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barseghyan, H.; Pang, A.W.C.; Clifford, B.; Serrano, M.A.; Chaubey, A.; Hastie, A.R. Comparative Benchmarking of Optical Genome Mapping and Chromosomal Microarray Reveals High Technological Concordance in CNV Identification and Additional Structural Variant Refinement. Genes 2023, 14, 1868. https://doi.org/10.3390/genes14101868
Barseghyan H, Pang AWC, Clifford B, Serrano MA, Chaubey A, Hastie AR. Comparative Benchmarking of Optical Genome Mapping and Chromosomal Microarray Reveals High Technological Concordance in CNV Identification and Additional Structural Variant Refinement. Genes. 2023; 14(10):1868. https://doi.org/10.3390/genes14101868
Chicago/Turabian StyleBarseghyan, Hayk, Andy Wing Chun Pang, Benjamin Clifford, Moises A. Serrano, Alka Chaubey, and Alex R. Hastie. 2023. "Comparative Benchmarking of Optical Genome Mapping and Chromosomal Microarray Reveals High Technological Concordance in CNV Identification and Additional Structural Variant Refinement" Genes 14, no. 10: 1868. https://doi.org/10.3390/genes14101868
APA StyleBarseghyan, H., Pang, A. W. C., Clifford, B., Serrano, M. A., Chaubey, A., & Hastie, A. R. (2023). Comparative Benchmarking of Optical Genome Mapping and Chromosomal Microarray Reveals High Technological Concordance in CNV Identification and Additional Structural Variant Refinement. Genes, 14(10), 1868. https://doi.org/10.3390/genes14101868