
Genome-Wide Association Study Reveals Additive and Non-Additive Effects on Growth Traits in Duroc Pigs

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Phenotype Data
2.2. Genotype Data and Quality Control
2.3. Statistical Analyses
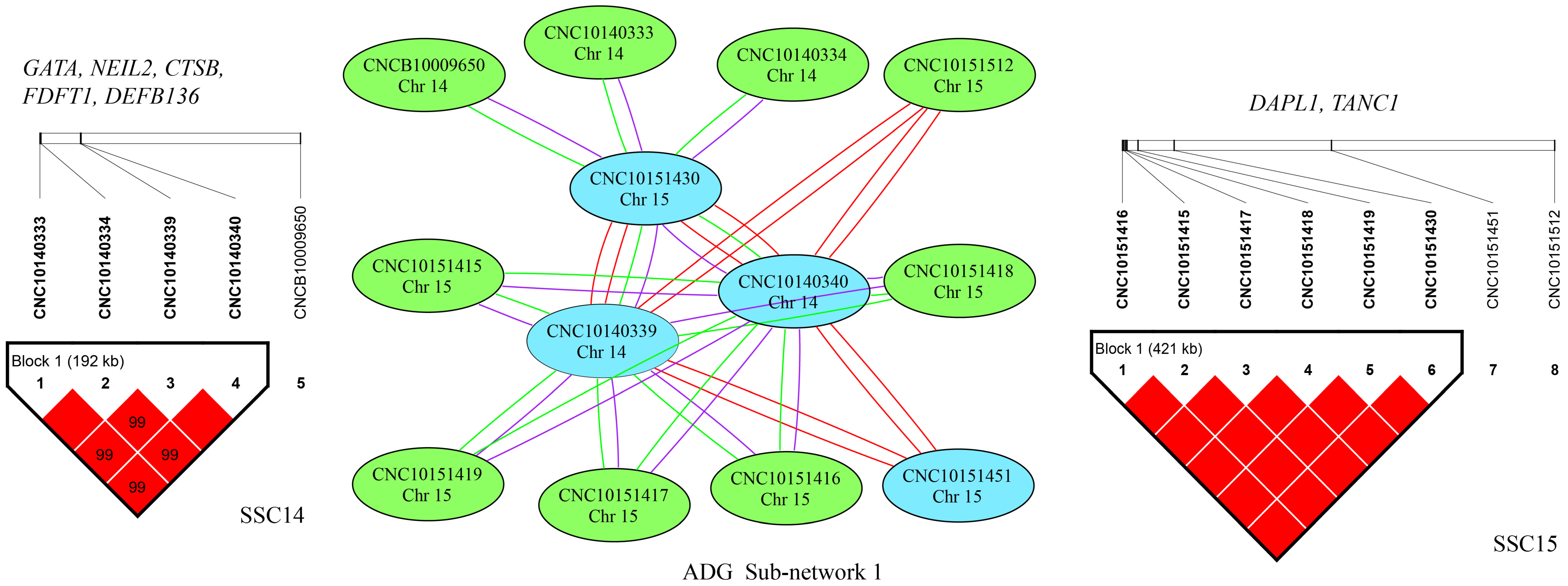
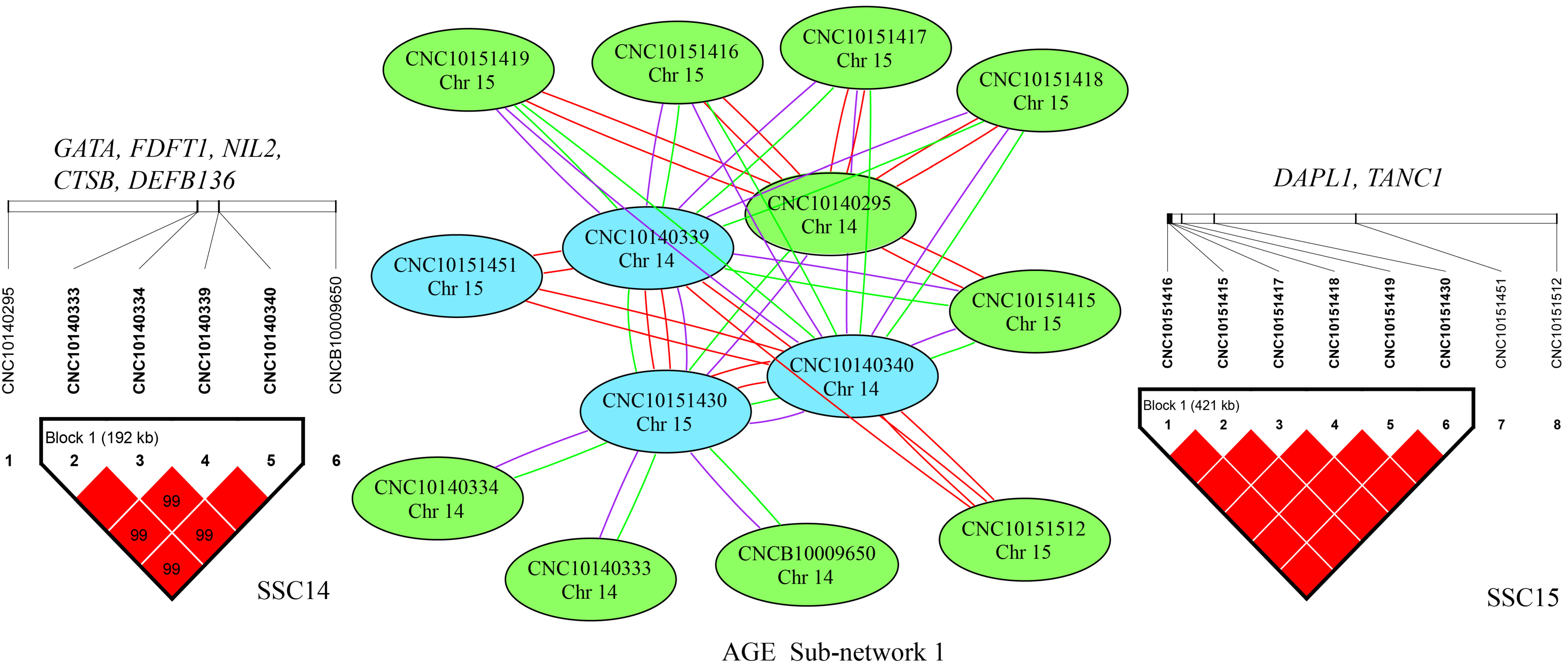
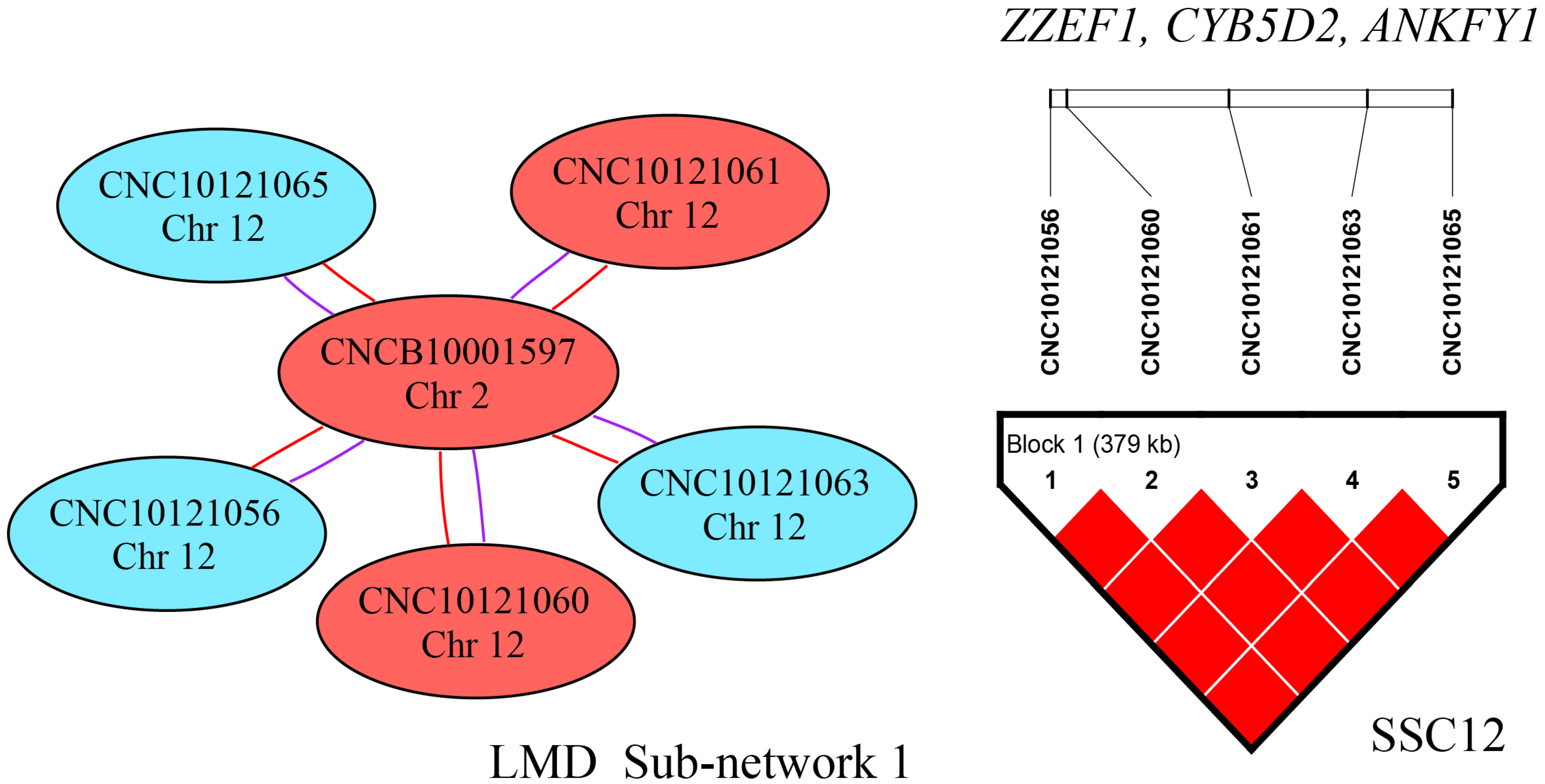
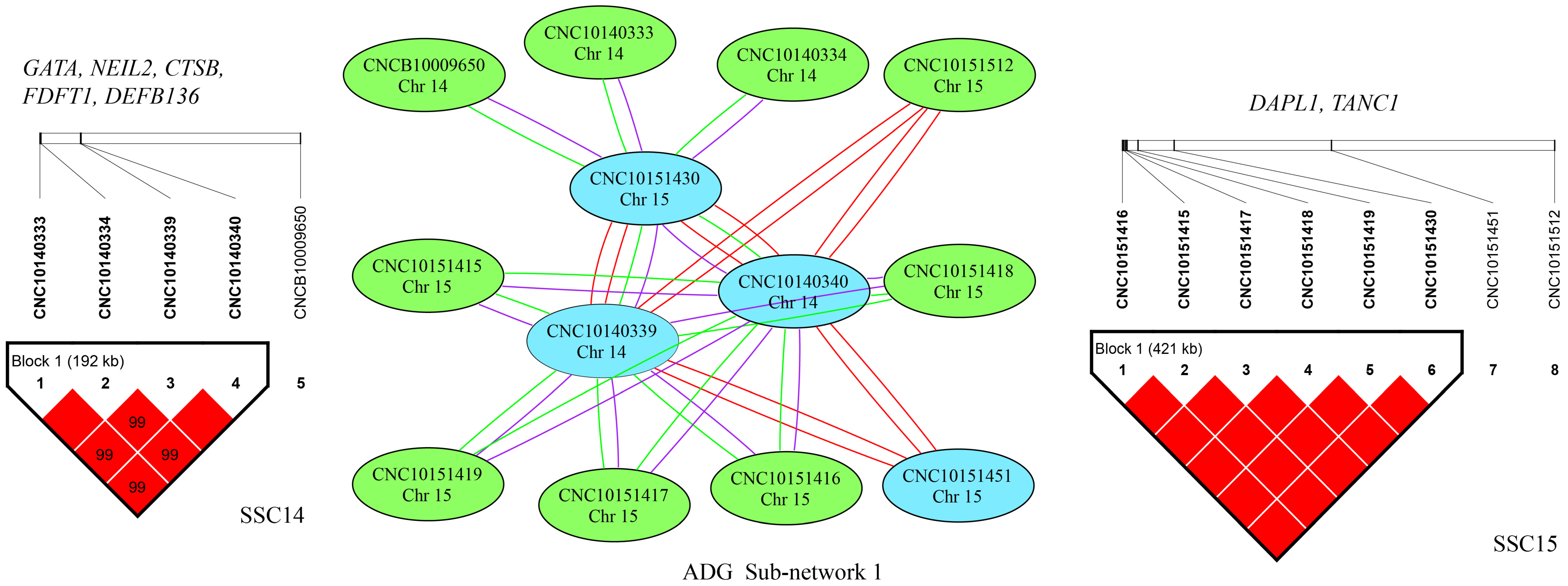
2.4. SNP–SNP Network and Linkage Disequilibrium (LD) Analysis
2.5. Identification of Candidate Genes
3. Results
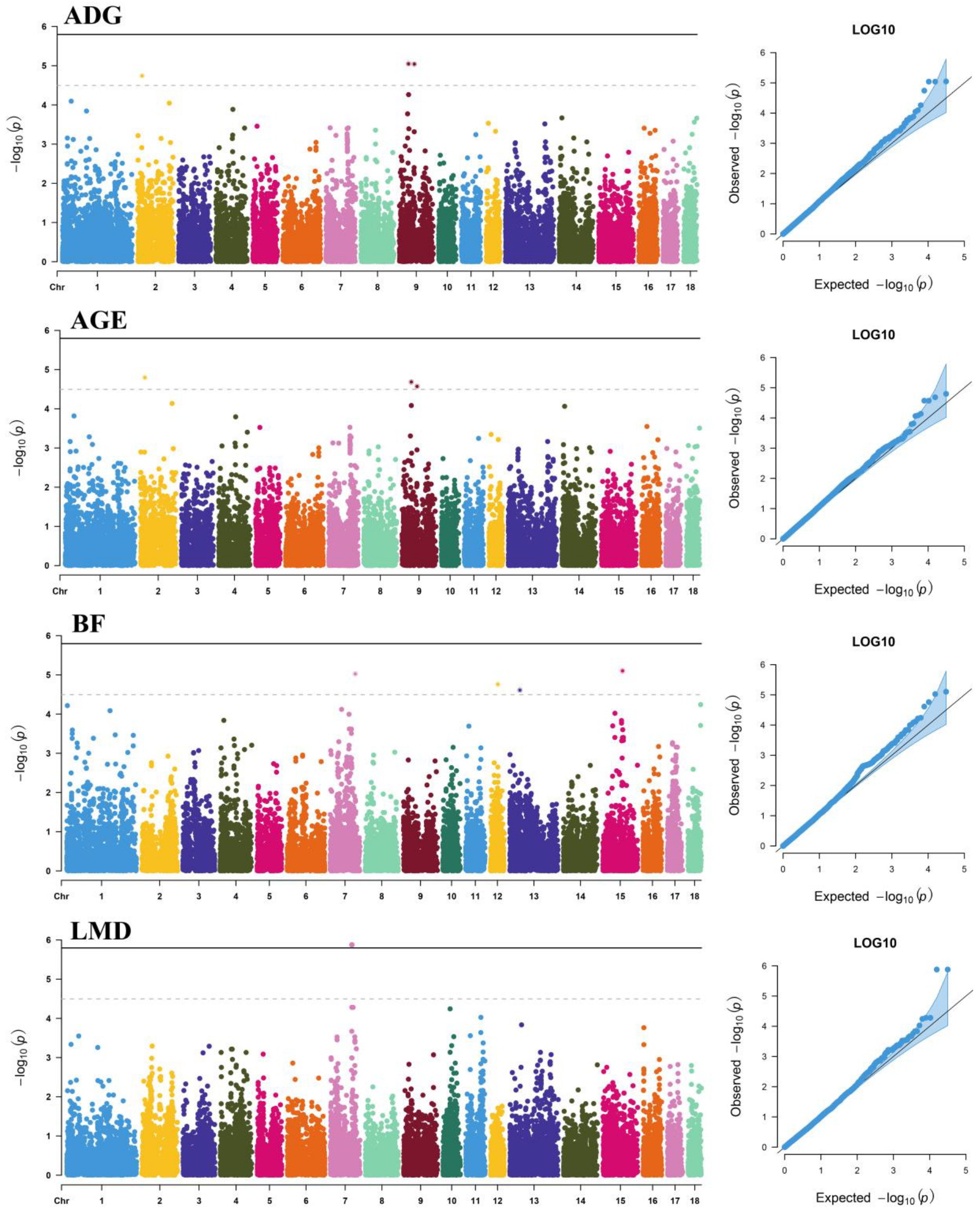
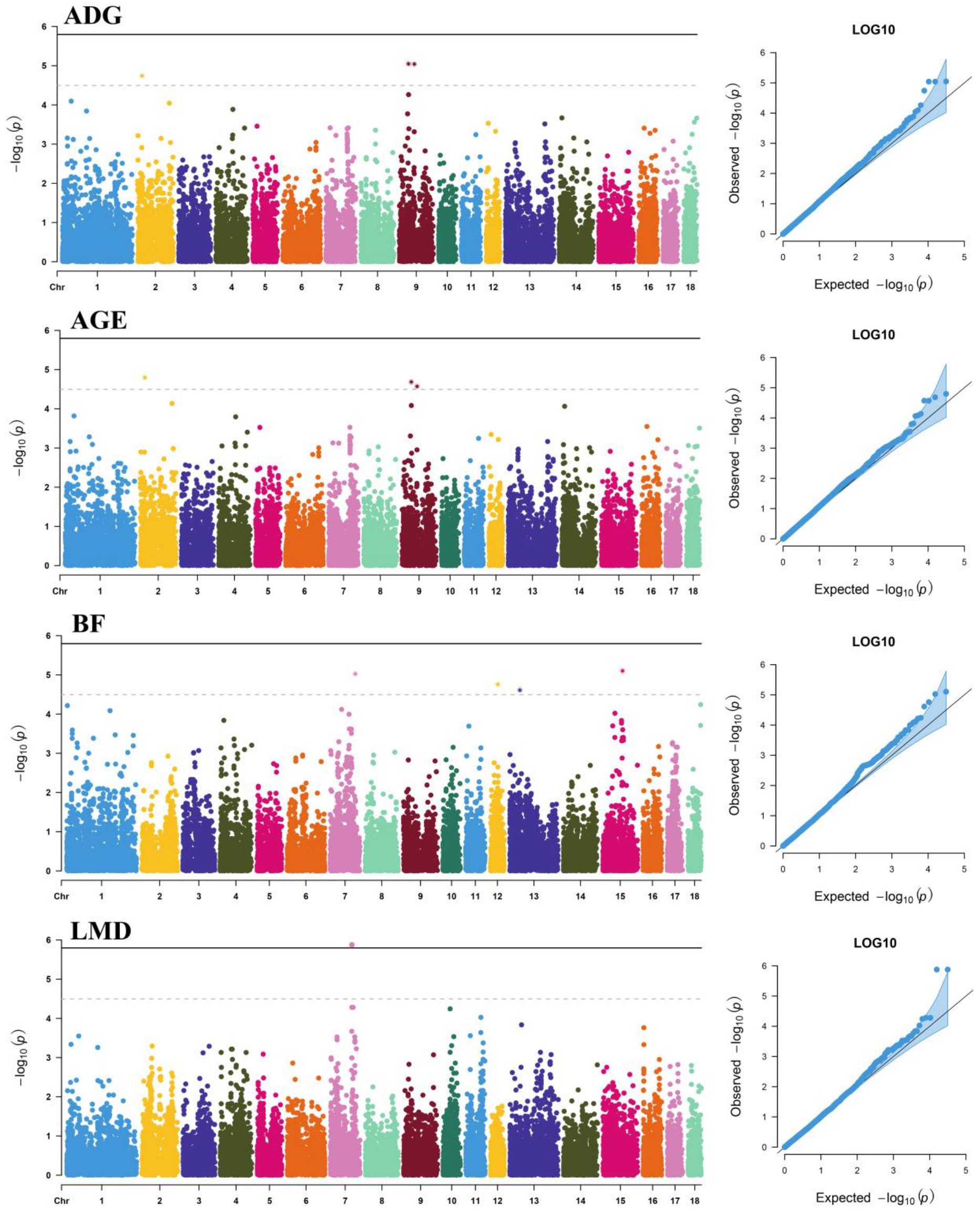
3.1. Additive Effects
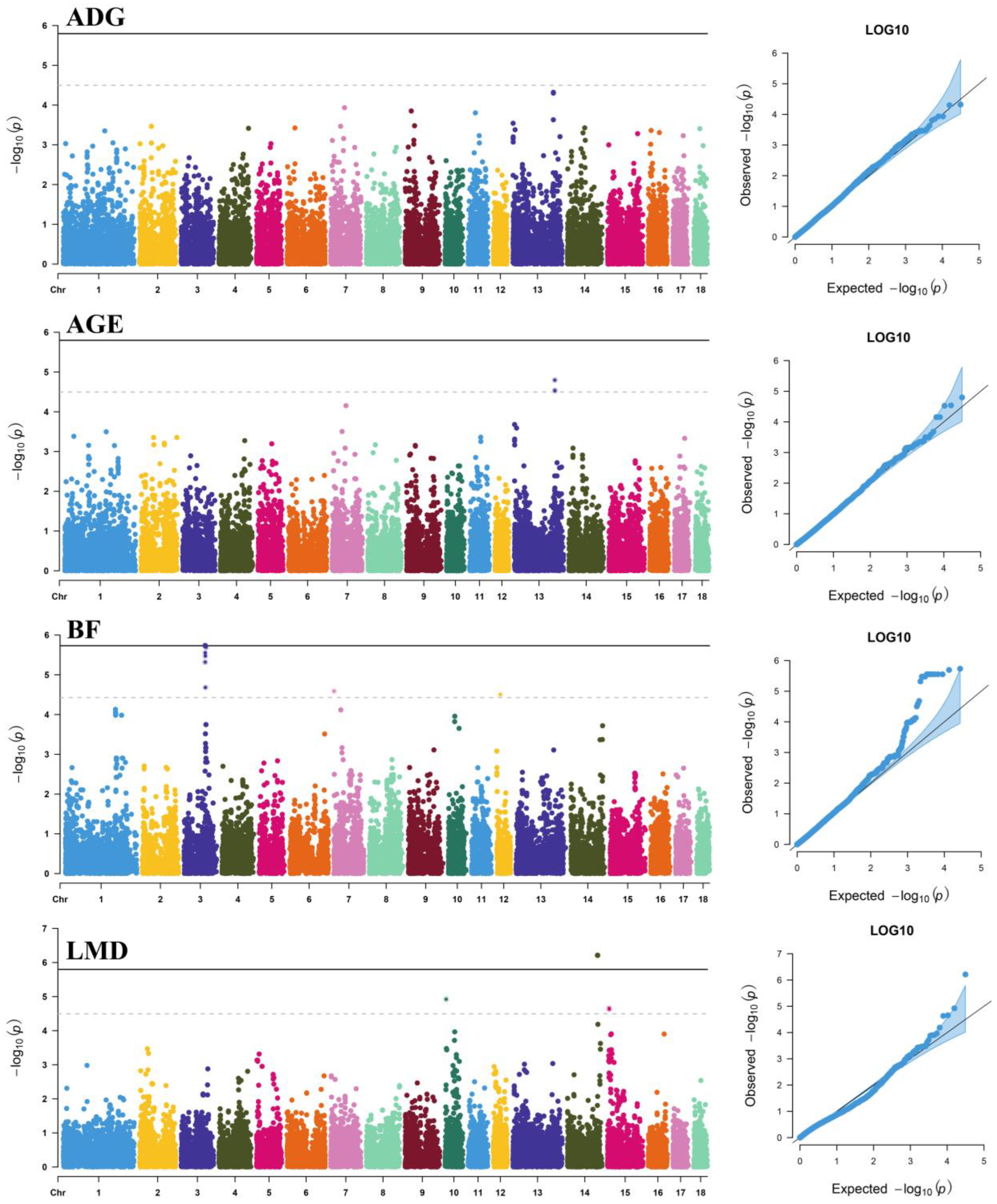
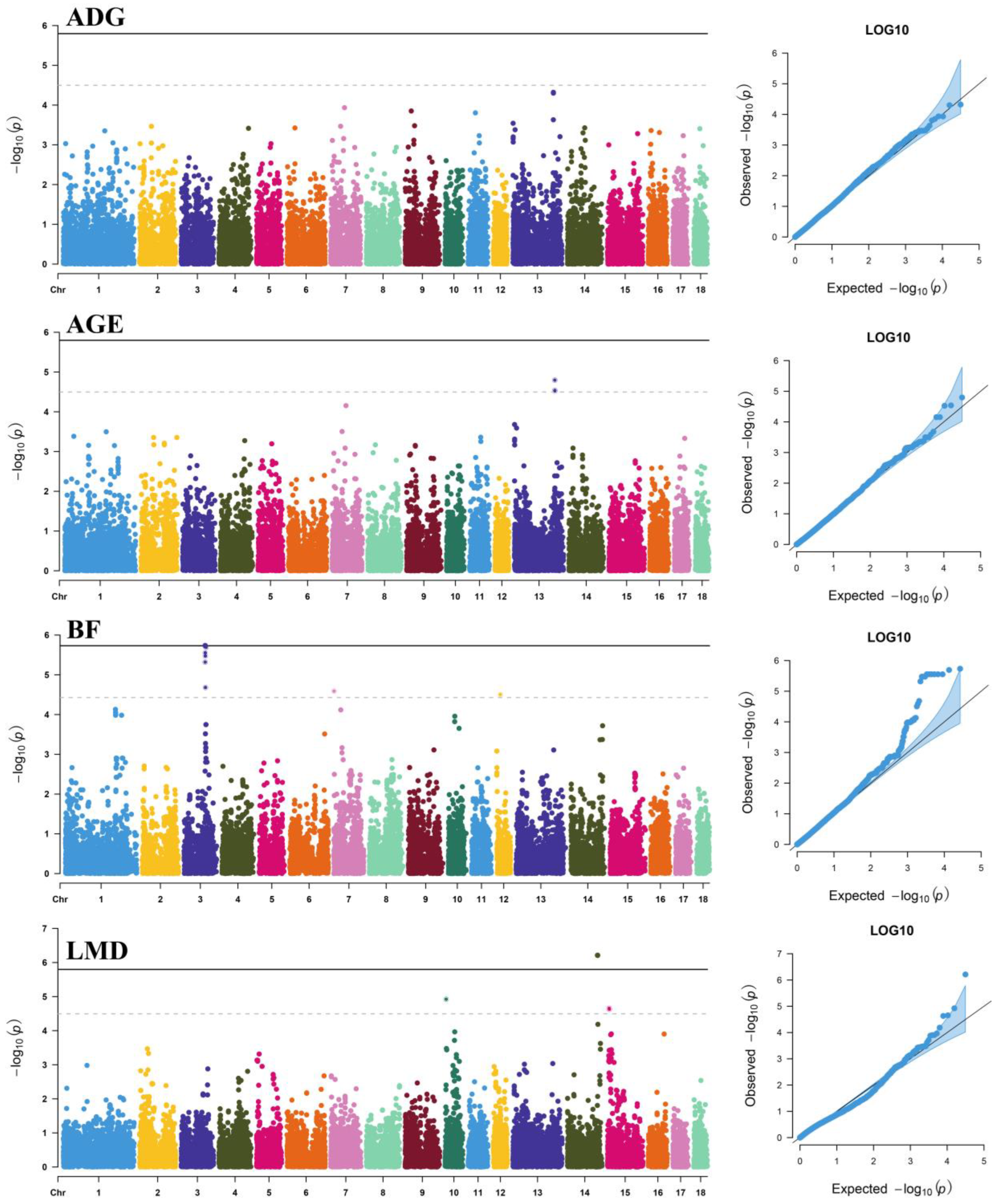
3.2. Dominance Effects
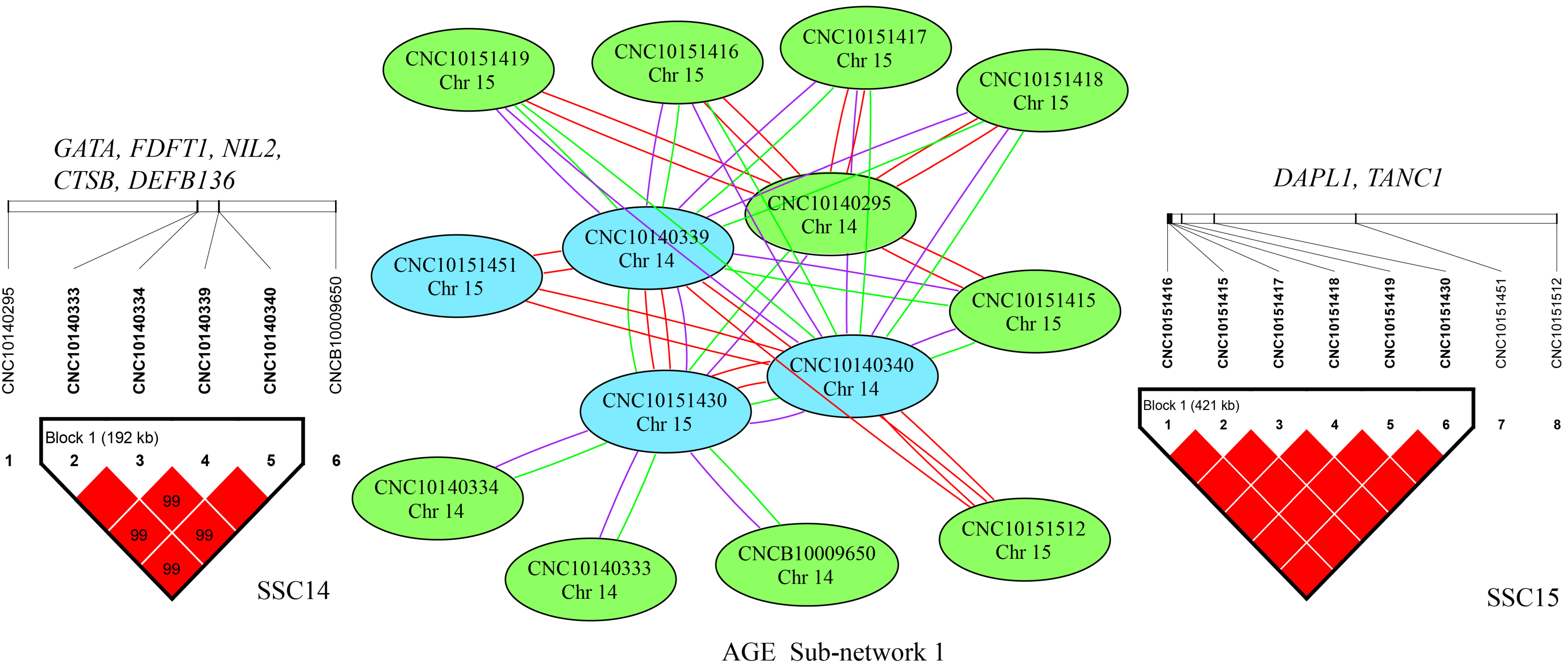
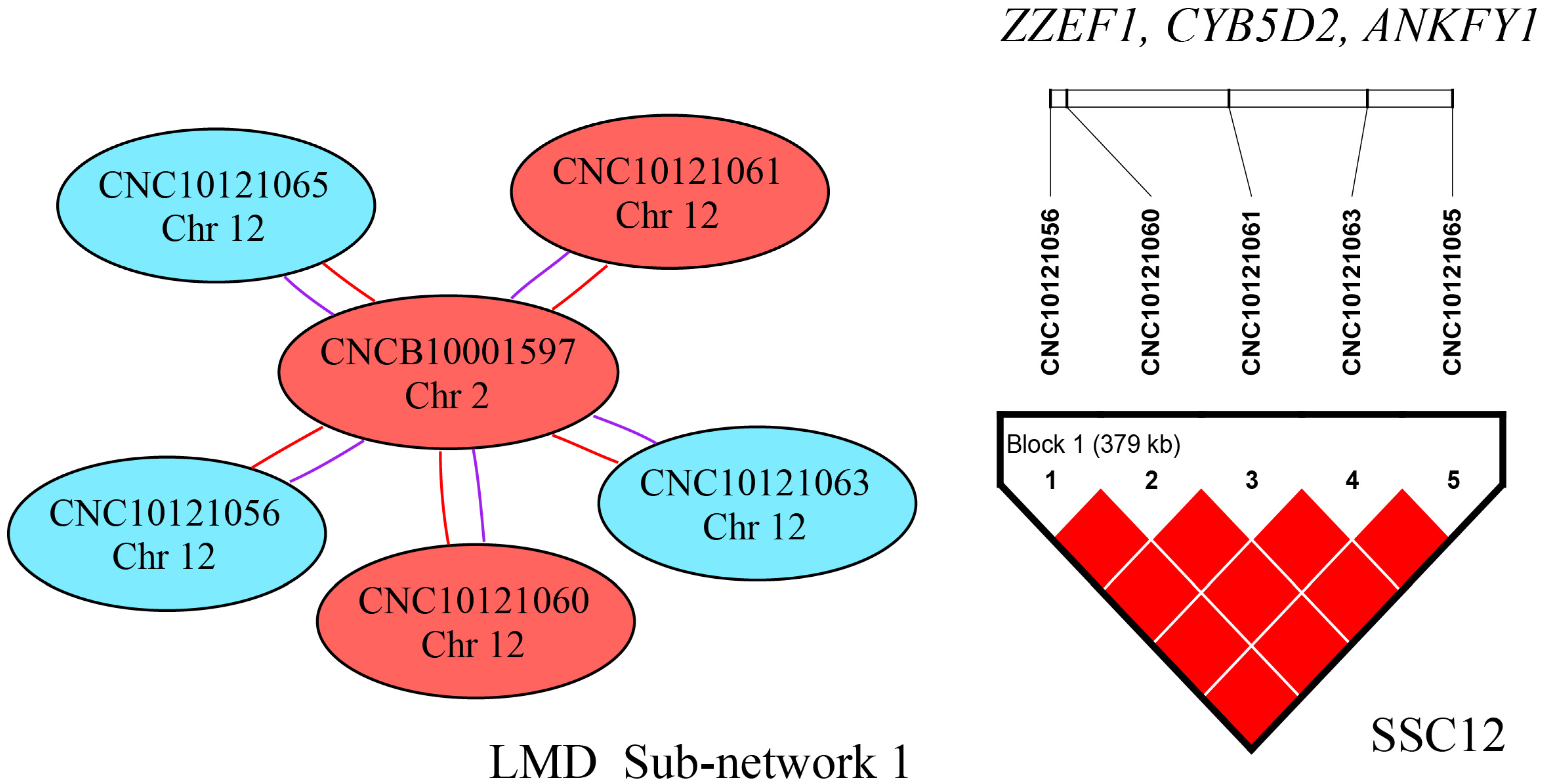
3.3. Epistatic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, K.; Liu, D.; Hernandez-Sanchez, J.; Chen, J.; Liu, C.; Wu, Z.; Fang, M.; Li, N. Genome wide association analysis reveals new production trait genes in a male Duroc population. PLoS ONE 2015, 10, e0139207. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Xu, J.; Yin, L.; Yin, D.; Zhu, M.; Yu, M.; Li, X.; Zhao, S.; Liu, X. Genome-wide association study reveals candidate genes for growth relevant traits in pigs. Front. Genet. 2019, 10, 302. [Google Scholar] [CrossRef]
- Hoque, M.A.; Suzuki, K.; Kadowaki, H.; Shibata, T.; Oikawa, T. Genetic parameters for feed efficiency traits and their relationships with growth and carcass traits in Duroc pigs. J. Anim. Breed. Genet. 2007, 124, 108–116. [Google Scholar] [CrossRef]
- Bolormaa, S.; Pryce, J.E.; Zhang, Y.; Reverter, A.; Barendse, W.; Hayes, B.J.; Goddard, M.E. Non-additive genetic variation in growth, carcass and fertility traits of beef cattle. Genet Sel. Evol. 2015, 47, 26. [Google Scholar] [CrossRef] [PubMed]
- Vitezica, Z.G.; Reverter, A.; Herring, W.; Legarra, A. Dominance and epistatic genetic variances for litter size in pigs using genomic models. Genet. Sel. Evol. 2018, 50, 71. [Google Scholar] [CrossRef] [PubMed]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, X.; Tan, Z.; Xing, K.; Yang, T.; Wang, Y.; Sun, D.; Wang, C. Genome-wide association study for reproductive traits in a Large White pig population. Anim. Genet. 2018, 49, 127–131. [Google Scholar] [CrossRef]
- Akanno, E.C.; Chen, L.; Abo-Ismail, M.K.; Crowley, J.J.; Wang, Z.; Li, C.; Basarab, J.A.; MacNeil, M.D.; Plastow, G.S. Genome-wide association scan for heterotic quantitative trait loci in multi-breed and crossbred beef cattle. Genet. Sel. Evol. 2018, 50, 48. [Google Scholar] [CrossRef]
- Zepeda-Batista, J.L.; Núñez-Domínguez, R.; Ramírez-Valverde, R.; Jahuey-Martínez, F.J.; Herrera-Ojeda, J.B.; Parra-Bracamonte, G.M. Discovering of Genomic Variations Associated to Growth Traits by GWAS in Braunvieh Cattle. Genes 2021, 12, 1666. [Google Scholar] [CrossRef]
- Duan, X.; An, B.; Du, L.; Chang, T.; Liang, M.; Yang, B.G.; Xu, L.; Zhang, L.; Li, J.; E, G.; et al. Genome-Wide Association Analysis of Growth Curve Parameters in Chinese Simmental Beef Cattle. Animals 2021, 11, 192. [Google Scholar] [CrossRef]
- Abousoliman, I.; Reyer, H.; Oster, M.; Murani, E.; Mohamed, I.; Wimmers, K. Genome-Wide Analysis for Early Growth-Related Traits of the Locally Adapted Egyptian Barki Sheep. Genes 2021, 12, 1243. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Jiang, Y.; Wang, C.; Mei, M.; Zhou, Z.; Jiang, Y.; Song, H.; Ding, X. A genome-wide association study on feed efficiency related traits in Landrace pigs. Front. Genet. 2020, 11, 692. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Wang, K.; Yang, Q.; Zhou, J.; Chen, D.; Liu, Y.; Ma, J.; Tang, Q.; Jin, L.; Xiao, W. Whole-genome re-sequencing association study for direct genetic effects and social genetic effects of six growth traits in Large White pigs. Sci. Rep. 2019, 9, 9667. [Google Scholar] [CrossRef]
- Ruan, D.; Zhuang, Z.; Ding, R.; Qiu, Y.; Zhou, S.; Wu, J.; Xu, C.; Hong, L.; Huang, S.; Zheng, E.; et al. Weighted Single-Step GWAS Identified Candidate Genes Associated with Growth Traits in a Duroc Pig Population. Genes 2021, 12, 117. [Google Scholar] [CrossRef]
- Su, G.; Christensen, O.F.; Ostersen, T.; Henryon, M.; Lund, M.S. Estimating additive and non-additive genetic variances and predicting genetic merits using genome-wide dense single nucleotide polymorphism markers. PLoS ONE 2012, 7, e45293. [Google Scholar] [CrossRef] [PubMed]
- Aliloo, H.; Pryce, J.E.; González-Recio, O.; Cocks, B.G.; Hayes, B.J. Accounting for dominance to improve genomic evaluations of dairy cows for fertility and milk production traits. Genet. Sel. Evol. 2016, 48, 8. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A one-penny imputed genome from next-generation reference panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef]
- Madsen, P.; Jensen, J.; Labouriau, R.; Christensen, O.F.; Sahana, G. DMU—A Package for Analyzing Multivariate Mixed Models in quantitative Genetics and Genomics. In Proceedings of the 10th World Congress of Genetics Applied to Livestock Production, Vancouver, BC, Canada, 17–22 August 2014; pp. 18–22. [Google Scholar]
- Garrick, D.J.; Taylor, J.F.; Fernando, R.L. Deregressing estimated breeding values and weighting information for genomic regression analyses. Genet. Sel. Evol. 2009, 41, 55. [Google Scholar] [CrossRef]
- Mei, Q.; Fu, C.; Li, J.; Zhao, S.; Xiang, T. blupADC: An R package and shiny toolkit for comprehensive genetic data analysis in animal and plant breeding. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wang, D.; Tang, H.; Liu, J.F.; Xu, S.; Zhang, Q.; Ning, C. Rapid epistatic mixed-model association studies by controlling multiple polygenic effects. Bioinformatics 2020, 36, 4833–4837. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.; Chasman, D.I.; Smith, J.D.; Mora, S.; Ridker, P.M.; Nickerson, D.A.; Krauss, R.M.; Stephens, M. A multivariate genome-wide association analysis of 10 LDL subfractions, and their response to statin treatment, in 1868 Caucasians. PLoS ONE 2015, 10, e0120758. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Runesha, H.B.; Dvorkin, D.; Garbe, J.R.; Da, Y. Parallel and serial computing tools for testing single-locus and epistatic SNP effects of quantitative traits in genome-wide association studies. BMC Bioinform. 2008, 9, 315. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef]
- Hu, Z.-L.; Park, C.A.; Reecy, J.M. Bringing the Animal QTLdb and CorrDB into the future: Meeting new challenges and providing updated services. Nucleic Acids Res. 2022, 50, D956–D961. [Google Scholar] [CrossRef]
- Safran, M.; Rosen, N.; Twik, M.; BarShir, R.; Stein, T.I.; Dahary, D.; Fishilevich, S.; Lancet, D. The GeneCards Suite. In Practical Guide to Life Science Databases; Abugessaisa, I., Kasukawa, T., Eds.; Springer Nature Singapore: Singapore, 2022; pp. 27–56. [Google Scholar]
- Coordinators, N.R. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, Z.; Ye, S.; He, Y.; Huang, S.; Yuan, X.; Chen, Z.; Zhang, H.; Li, J. Genome-Wide Association Study for Reproductive Traits in a Duroc Pig Population. Animals 2019, 9, 732. [Google Scholar] [CrossRef]
- Lee, J.; Kang, J.H.; Kim, J.M. Bayes Factor-Based Regulatory Gene Network Analysis of Genome-Wide Association Study of Economic Traits in a Purebred Swine Population. Genes 2019, 10, 293. [Google Scholar] [CrossRef]
- Sutera, A.M.; Moscarelli, A.; Mastrangelo, S.; Sardina, M.T.; Di Gerlando, R.; Portolano, B.; Tolone, M. Genome-Wide Association Study Identifies New Candidate Markers for Somatic Cells Score in a Local Dairy Sheep. Front. Genet. 2021, 12, 643531. [Google Scholar] [CrossRef]
- Zhang, H.; Yu, J.-Q.; Yang, L.-L.; Kramer, L.M.; Zhang, X.-Y.; Na, W.; Reecy, J.M.; Li, H. Identification of genome-wide SNP-SNP interactions associated with important traits in chicken. BMC Genom. 2017, 18, 892. [Google Scholar] [CrossRef] [PubMed]
- Kramer, L.M.; Ghaffar, M.A.; Koltes, J.E.; Fritz-Waters, E.R.; Mayes, M.S.; Sewell, A.D.; Weeks, N.T.; Garrick, D.J.; Fernando, R.L.; Ma, L.; et al. Epistatic interactions associated with fatty acid concentrations of beef from angus sired beef cattle. BMC Genom. 2016, 17, 891. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Wang, K.; Liu, X.; Zhou, H.; Xu, L.; Wang, Z.; Fang, M. Identification of growth trait related genes in a Yorkshire purebred pig population by genome-wide association studies. Asian-Australas. J. Anim. Sci. 2017, 30, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Gronthos, S.; Franklin, D.M.; Leddy, H.A.; Robey, P.G.; Storms, R.W.; Gimble, J.M. Surface protein characterization of human adipose tissue-derived stromal cells. J. Cell. Physiol. 2001, 189, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Festy, F.; Hoareau, L.; Bes-Houtmann, S.; Péquin, A.-M.; Gonthier, M.-P.; Munstun, A.; Hoarau, J.J.; Césari, M.; Roche, R. Surface protein expression between human adipose tissue-derived stromal cells and mature adipocytes. Histochem. Cell Biol. 2005, 124, 113–121. [Google Scholar] [CrossRef]
- Christian, M.; Kiskinis, E.; Debevec, D.; Leonardsson, G.; White, R.; Parker, M.G. RIP140-targeted repression of gene expression in adipocytes. Mol. Cell Biol. 2005, 25, 9383–9391. [Google Scholar] [CrossRef]
- Ho, P.C.; Chuang, Y.S.; Hung, C.H.; Wei, L.N. Cytoplasmic receptor-interacting protein 140 (RIP140) interacts with perilipin to regulate lipolysis. Cell Signal. 2011, 23, 1396–1403. [Google Scholar] [CrossRef]
- Hochberg, I.; Tran, Q.T.; Barkan, A.L.; Saltiel, A.R.; Chandler, W.F.; Bridges, D. Gene Expression Signature in Adipose Tissue of Acromegaly Patients. PLoS ONE 2015, 10, e0129359. [Google Scholar] [CrossRef]
- Shen, Y.; Cohen, J.L.; Nicoloro, S.M.; Kelly, M.; Yenilmez, B.; Henriques, F.; Tsagkaraki, E.; Edwards, Y.J.K.; Hu, X.; Friedline, R.H.; et al. CRISPR-delivery particles targeting nuclear receptor-interacting protein 1 (Nrip1) in adipose cells to enhance energy expenditure. J. Biol. Chem. 2018, 293, 17291–17305. [Google Scholar] [CrossRef]
- Saatcioglu, H.D.; Kano, M.; Horn, H.; Zhang, L.; Samore, W.; Nagykery, N.; Meinsohn, M.C.; Hyun, M.; Suliman, R.; Poulo, J.; et al. Single-cell sequencing of neonatal uterus reveals an Misr2+ endometrial progenitor indispensable for fertility. eLife 2019, 8, e46349. [Google Scholar] [CrossRef]
- Verardo, L.L.; Silva, F.F.; Lopes, M.S.; Madsen, O.; Bastiaansen, J.W.; Knol, E.F.; Kelly, M.; Varona, L.; Lopes, P.S.; Guimarães, S.E. Revealing new candidate genes for reproductive traits in pigs: Combining Bayesian GWAS and functional pathways. Genet. Sel. Evol. 2016, 48, 9. [Google Scholar] [CrossRef] [PubMed]
- Gurgul, A.; Jasielczuk, I.; Ropka-Molik, K.; Semik-Gurgul, E.; Pawlina-Tyszko, K.; Szmatoła, T.; Szyndler-Nędza, M.; Bugno-Poniewierska, M.; Blicharski, T.; Szulc, K.; et al. A genome-wide detection of selection signatures in conserved and commercial pig breeds maintained in Poland. BMC Genet. 2018, 19, 95. [Google Scholar] [CrossRef] [PubMed]
- Ayuso, M.; Fernández, A.; Núñez, Y.; Benítez, R.; Isabel, B.; Barragán, C.; Fernández, A.I.; Rey, A.I.; Medrano, J.F.; Cánovas, Á.; et al. Comparative Analysis of Muscle Transcriptome between Pig Genotypes Identifies Genes and Regulatory Mechanisms Associated to Growth, Fatness and Metabolism. PLoS ONE 2015, 10, e0145162. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Cheng, S.Y. New insights into regulation of lipid metabolism by thyroid hormone. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Hasebe, H.; Sato, S.; Asahi, Y.; Hayashi, T.; Kobayashi, E.; Sugimoto, Y. High-resolution physical mapping and construction of a porcine contig spanning the intramuscular fat content QTL. Anim. Genet. 2006, 37, 113–120. [Google Scholar] [CrossRef]
- Do, D.N.; Strathe, A.B.; Ostersen, T.; Jensen, J.; Mark, T.; Kadarmideen, H.N. Genome-Wide Association Study Reveals Genetic Architecture of Eating Behavior in Pigs and Its Implications for Humans Obesity by Comparative Mapping. PLoS ONE 2013, 8, e71509. [Google Scholar] [CrossRef]
- Sakakibara, S.; Nakamura, Y.; Satoh, H.; Okano, H. Rna-binding protein Musashi2: Developmentally regulated expression in neural precursor cells and subpopulations of neurons in mammalian CNS. J. Neurosci. 2001, 21, 8091–8107. [Google Scholar] [CrossRef]
- Fowler, K.E.; Pong-Wong, R.; Bauer, J.; Clemente, E.J.; Reitter, C.P.; Affara, N.A.; Waite, S.; Walling, G.A.; Griffin, D.K. Genome wide analysis reveals single nucleotide polymorphisms associated with fatness and putative novel copy number variants in three pig breeds. BMC Genom. 2013, 14, 784. [Google Scholar] [CrossRef]
- Nakano, H.; Sato, S.; Uemoto, Y.; Kikuchi, T.; Shibata, T.; Kadowaki, H.; Kobayashi, E.; Suzuki, K. Effect of VRTN gene polymorphisms on Duroc pig production and carcass traits, and their genetic relationships. Anim. Sci. J. 2015, 86, 125–131. [Google Scholar] [CrossRef]
- Hirose, K.; Mikawa, S.; Okumura, N.; Noguchi, G.; Fukawa, K.; Kanaya, N.; Mikawa, A.; Arakawa, A.; Ito, T.; Hayashi, Y.; et al. Association of swine vertnin (VRTN) gene with production traits in Duroc pigs improved using a closed nucleus breeding system. Anim. Sci. J. 2013, 84, 213–221. [Google Scholar] [CrossRef]
- Di Filippo, E.S.; Costamagna, D.; Giacomazzi, G.; Cortés-Calabuig, Á.; Stryjewska, A.; Huylebroeck, D.; Fulle, S.; Sampaolesi, M. Zeb2 Regulates Myogenic Differentiation in Pluripotent Stem Cells. Int. J. Mol. Sci. 2020, 21, 2525. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Mean ± SD | Max. | Min. | C.V. (%) | Number of Records |
|---|---|---|---|---|---|
| ADG (g/day) | 661.66 ± 57.52 | 824.94 | 512.03 | 9.10 | 1854 |
| AGE (day) | 152.33 ± 13.86 | 195.30 | 121.22 | 17.67 | 1854 |
| BF (mm) | 9.58 ± 1.69 | 15.54 | 5.70 | 8.69 | 1847 |
| LMD (mm) | 54.87 ± 6.52 | 74.01 | 27.35 | 11.89 | 1651 |
| Trait | SNP | Chr | Location (bp) | Allele | MAF | p-Value | % DEBV | Nearest Gene | Distance (bp) |
|---|---|---|---|---|---|---|---|---|---|
| ADG | CNCB10006667 | 9 | 41,538,619 | C/T | 0.086 | 8.95 × 10−6 | 0.83 | HTR3B | Within |
| CNCB10006791 | 9 | 67,900,715 | C/T | 0.086 | 9.09 × 10−6 | 0.83 | CD55 | +32,354 | |
| CNCB10006792 | 9 | 67,967,237 | A/G | 0.086 | 9.09 × 10−6 | 0.83 | CD55 | −6855 | |
| AGE | CNCB10001620 | 2 | 20,310,474 | A/G | 0.216 | 1.59 × 10−5 | 0.78 | / | / |
| CNCB10006667 | 9 | 41,538,619 | C/T | 0.086 | 2.07 × 10−5 | 0.76 | HTR3B | Within | |
| CNCB10006791 | 9 | 67,900,715 | C/T | 0.086 | 2.68 × 10−5 | 0.74 | CD55 | +32,354 | |
| CNCB10006792 | 9 | 67,967,237 | A/G | 0.086 | 2.68 × 10−5 | 0.74 | CD55 | −6855 | |
| BF | CNCB10010792 | 15 | 89,052,971 | G/A | 0.051 | 7.89 × 10−6 | 0.84 | / | / |
| CNCB10005591 | 7 | 113,533,476 | T/G | 0.074 | 9.44 × 10−6 | 0.82 | TRIP11 | Within | |
| rs81433919 | 12 | 33,673,968 | G/A | 0.116 | 1.74 × 10−5 | 0.77 | MSI2 | Within | |
| CNCB10008592 | 13 | 45,621,675 | G/A | 0.072 | 2.45 × 10−5 | 0.75 | PRICKLE2 | Within | |
| LMD | rs709317845 | 7 | 97,614,602 | T/G | 0.469 | 1.33 × 10−6 | 0.98 | VRTN | +105 |
| CNC11071978 | 7 | 97,615,897 | A/G | 0.469 | 1.33 × 10−6 | 0.98 | VRTN | Within |
| Trait | SNP | Chr | Location (bp) | Allele | MAF | p-Value | % DEBV | Nearest Gene | Distance (bp) |
|---|---|---|---|---|---|---|---|---|---|
| AGE | CNC10133718 | 13 | 179,900,741 | C/T | 0.285 | 1.60 × 10−5 | 0.78 | NRIP1 | Within |
| rs81441574 | 13 | 180,051,342 | T/C | 0.364 | 2.90 × 10−5 | 0.73 | NRIP1 | −135,116 | |
| rs80867343 | 13 | 180,004,272 | G/A | 0.362 | 2.98 × 10−5 | 0.73 | NRIP1 | −88,046 | |
| BF | rs81373550 | 3 | 93,497,234 | A/G | 0.061 | 1.85 × 10−6 | 0.95 | TTC7A | Within |
| CNCB10002800 | 3 | 97,223,093 | T/C | 0.149 | 2.03 × 10−6 | 0.95 | ZFP36L2 | +19,999 | |
| rs81373610 | 3 | 93,655,118 | A/C | 0.060 | 2.80 × 10−6 | 0.92 | MCFD2 | +22,100 | |
| rs80966590 | 3 | 93,722,940 | C/T | 0.060 | 2.80 × 10−6 | 0.92 | SOCS5 | −54,462 | |
| rs81475091 | 3 | 93,732,314 | G/A | 0.060 | 2.80 × 10−6 | 0.92 | SOCS5 | −45,088 | |
| rs81299554 | 3 | 93,771,724 | A/G | 0.060 | 2.80 × 10−6 | 0.92 | SOCS5 | −5678 | |
| rs81373642 | 3 | 93,828,846 | C/T | 0.060 | 2.80 × 10−6 | 0.92 | SOCS5 | Within | |
| rs81373648 | 3 | 93,902,023 | G/T | 0.060 | 2.80 × 10−6 | 0.92 | SOCS5 | Within | |
| rs81373727 | 3 | 94,084,767 | A/G | 0.060 | 3.36 × 10−6 | 0.91 | TMEM247 | +17,743 | |
| rs81373716 | 3 | 94,110,331 | G/A | 0.060 | 3.36 × 10−6 | 0.91 | TMEM247 | +43,307 | |
| rs81373744 | 3 | 94,163,788 | G/A | 0.060 | 3.36 × 10−6 | 0.91 | EPAS1 | −3971 | |
| rs81213041 | 3 | 93,394,317 | G/A | 0.059 | 4.80 × 10−6 | 0.88 | STPG4 | Within | |
| rs81373880 | 3 | 94,599,697 | C/T | 0.075 | 2.09 × 10−5 | 0.76 | PRKCE | Within | |
| CNC10070016 | 7 | 834,427 | A/C | 0.430 | 2.57 × 10−5 | 0.74 | GMDS | Within | |
| CNC10120338 | 12 | 16,082,351 | T/C | 0.488 | 3.13 × 10−5 | 0.73 | MRC2 | Within | |
| LMD | rs80785395 | 14 | 133,966,414 | A/G | 0.087 | 6.14 × 10−7 | 1.04 | LHPP | Within |
| CNCB10007305 | 10 | 4,667,593 | A/G | 0.094 | 1.19 × 10−5 | 0.80 | / | / | |
| rs81304718 | 15 | 7,424,079 | C/T | 0.091 | 2.21 × 10−5 | 0.76 | ZEB2 | −74,800 | |
| CNCB10010367 | 15 | 8,203,810 | A/G | 0.060 | 2.34 × 10−5 | 0.75 | ARHGAP15 | Within |
| Traits | N | A × A | A × D | D × D |
|---|---|---|---|---|
| ADG | 287 | 124 | 97 | 66 |
| AGE | 355 | 159 | 135 | 61 |
| LMD | 163 | 80 | 53 | 30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, Y.; Liu, S.; Li, W.; Mao, R.; Zhuo, Y.; Xing, W.; Liu, J.; Wang, C.; Zhou, L.; Lei, M.; et al. Genome-Wide Association Study Reveals Additive and Non-Additive Effects on Growth Traits in Duroc Pigs. Genes 2022, 13, 1454. https://doi.org/10.3390/genes13081454
Xue Y, Liu S, Li W, Mao R, Zhuo Y, Xing W, Liu J, Wang C, Zhou L, Lei M, et al. Genome-Wide Association Study Reveals Additive and Non-Additive Effects on Growth Traits in Duroc Pigs. Genes. 2022; 13(8):1454. https://doi.org/10.3390/genes13081454
Chicago/Turabian StyleXue, Yahui, Shen Liu, Weining Li, Ruihan Mao, Yue Zhuo, Wenkai Xing, Jian Liu, Chuang Wang, Lei Zhou, Minggang Lei, and et al. 2022. "Genome-Wide Association Study Reveals Additive and Non-Additive Effects on Growth Traits in Duroc Pigs" Genes 13, no. 8: 1454. https://doi.org/10.3390/genes13081454
APA StyleXue, Y., Liu, S., Li, W., Mao, R., Zhuo, Y., Xing, W., Liu, J., Wang, C., Zhou, L., Lei, M., & Liu, J. (2022). Genome-Wide Association Study Reveals Additive and Non-Additive Effects on Growth Traits in Duroc Pigs. Genes, 13(8), 1454. https://doi.org/10.3390/genes13081454