
Large Fragment InDels Reshape Genome Structure of Porcine Alveolar Macrophage 3D4/21 Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. All-Seq Library Preparation
2.2. Cell Culture and PAMs Collection
2.3. DNA Extraction
2.4. Identification of Large InDels
2.5. InDels Validation by PCR and Sanger Sequencing
2.6. RNA-Seq
2.7. Hi-C
3. Results
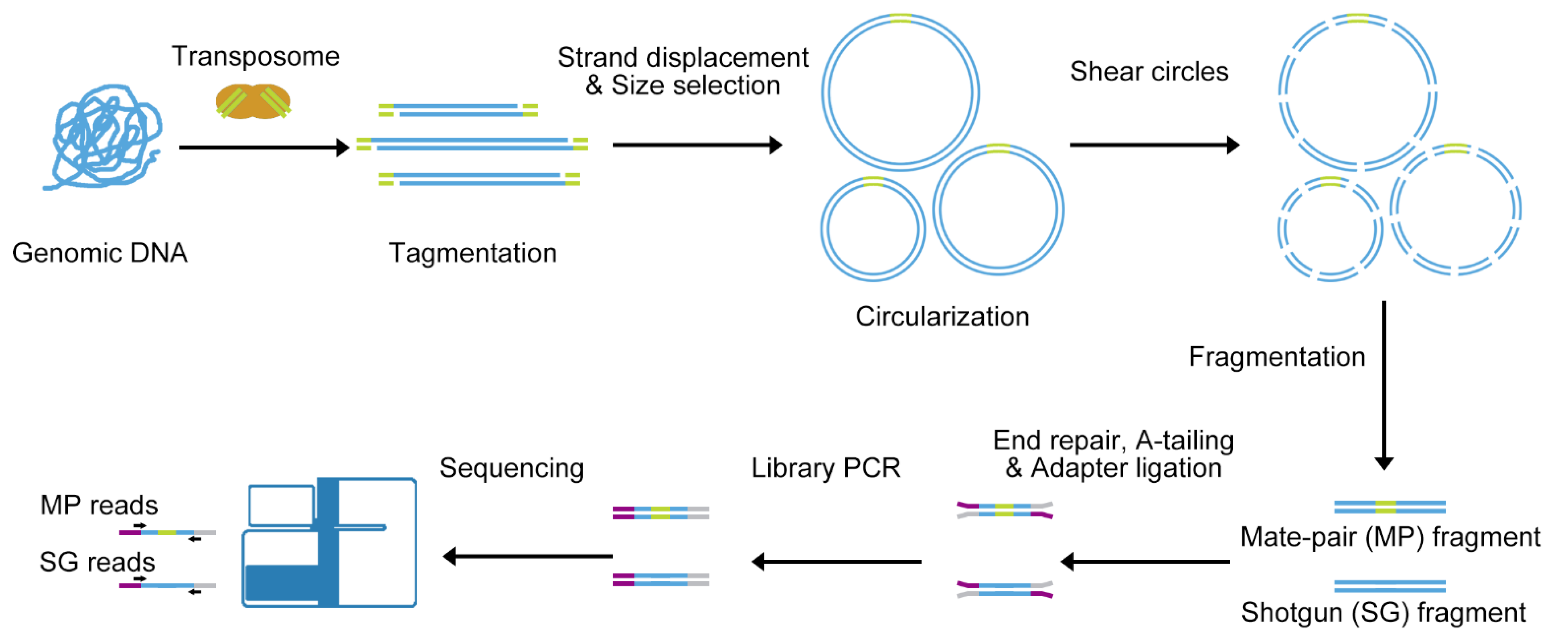
3.1. All-Seq Methodology
3.2. Quality Identification of All-Seq Library
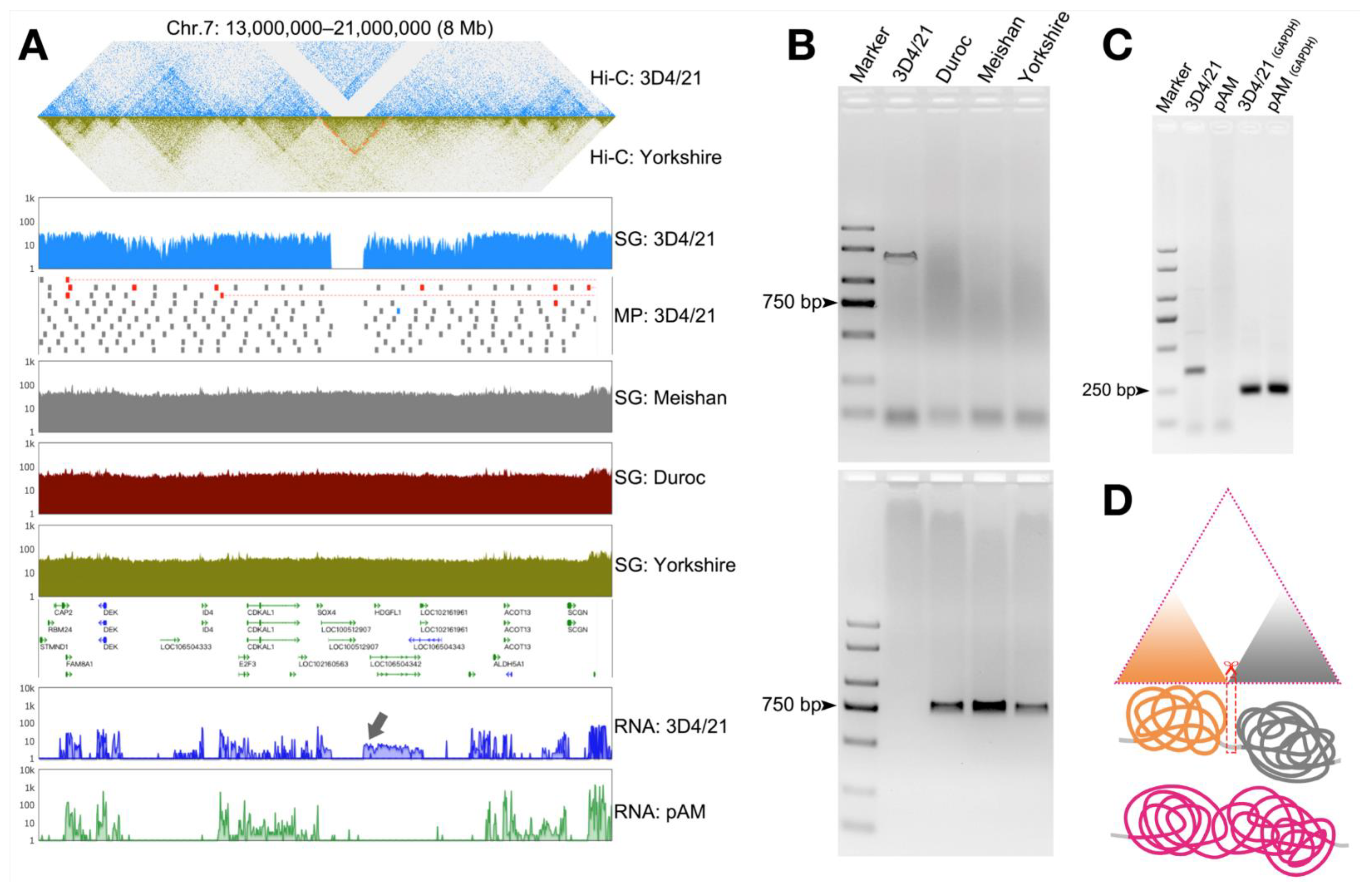
3.3. Detection of Structural Variation in 3D4/21 Cells
3.4. Validation of Structural Variation in 3D4/21 Cells
3.5. Structural Variation in the Genome Can Cause Changes in the Three-Dimensional Structure of Chromatin and Thus Affect Gene Transcription
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bendixen, E.; Danielsen, M.; Larsen, K.; Bendixen, C. Advances in porcine genomics and proteomics—A toolbox for developing the pig as a model organism for molecular biomedical research. Brief. Funct. Genom. 2010, 9, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Mou, Y.; Huang, Z.; Li, Y.; Han, L.; Zhang, Y.; Feng, Y.; Chen, Y.; Jiang, X.; Zhao, W.; et al. The sequence and analysis of a Chinese pig genome. Gigascience 2012, 1, 16. [Google Scholar] [CrossRef] [PubMed]
- Fan, N.; Lai, L. Genetically modified pig models for human diseases. J. Genet. Genom. 2013, 40, 67–73. [Google Scholar] [CrossRef]
- Prather, R.S.; Lorson, M.; Ross, J.W.; Whyte, J.J.; Walters, E. Genetically engineered pig models for human diseases. Annu. Rev. Anim. Biosci. 2013, 1, 203–219. [Google Scholar] [CrossRef]
- Yan, S.; Tu, Z.; Liu, Z.; Fan, N.; Yang, H.; Yang, S.; Yang, W.; Zhao, Y.; Ouyang, Z.; Lai, C.; et al. A Huntingtin Knockin Pig Model Recapitulates Features of Selective Neurodegeneration in Huntington’s Disease. Cell 2018, 173, 989–1002. [Google Scholar] [CrossRef]
- Itoh, M.; Mukae, Y.; Kitsuka, T.; Arai, K.; Nakamura, A.; Uchihashi, K.; Toda, S.; Matsubayashi, K.; Oyama, J.I.; Node, K.; et al. Development of an immunodeficient pig model allowing long-term accommodation of artificial human vascular tubes. Nat. Commun. 2019, 10, 2244. [Google Scholar] [CrossRef]
- Tanihara, F.; Takemoto, T.; Kitagawa, E.; Rao, S.; Do, L.T.; Onishi, A.; Yamashita, Y.; Kosugi, C.; Suzuki, H.; Sembon, S.; et al. Somatic cell reprogramming-free generation of genetically modified pigs. Sci. Adv. 2016, 2, e1600803. [Google Scholar] [CrossRef]
- Denner, J. Advances in organ transplant from pigs. Science 2017, 357, 1238–1239. [Google Scholar] [CrossRef] [PubMed]
- Porrett, P.M.; Orandi, B.J.; Kumar, V.; Houp, J.; Anderson, D.; Cozette Killian, A.; Hauptfeld-Dolejsek, V.; Martin, D.E.; Macedon, S.; Budd, N.; et al. First clinical-grade porcine kidney xenotransplant using a human decedent model. Am. J. Transplant. 2022, 22, 1037–1053. [Google Scholar] [CrossRef]
- Hussell, T.; Bell, T.J. Alveolar macrophages: Plasticity in a tissue-specific context. Nat. Rev. Immunol. 2014, 14, 81–93. [Google Scholar] [CrossRef]
- Morales-Nebreda, L.; Misharin, A.V.; Perlman, H.; Budinger, G.R. The heterogeneity of lung macrophages in the susceptibility to disease. Eur. Respir. Rev. 2015, 24, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.P.; Jacqueline, C.; Poschmann, J.; Roquilly, A. Alveolar Macrophages: Adaptation to Their Anatomic Niche during and after Inflammation. Cells 2021, 10, 2720. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Huang, K.; Liu, J.; Wu, S.; Shen, D.; Dai, P.; Li, C. Fine particulate matter from pig house induced immune response by activating TLR4/MAPK/NF-kappaB pathway and NLRP3 inflammasome in alveolar macrophages. Chemosphere 2019, 236, 124373. [Google Scholar] [CrossRef]
- Neupane, A.S.; Willson, M.; Chojnacki, A.K.; Vargas, E.S.C.F.; Morehouse, C.; Carestia, A.; Keller, A.E.; Peiseler, M.; DiGiandomenico, A.; Kelly, M.M.; et al. Patrolling Alveolar Macrophages Conceal Bacteria from the Immune System to Maintain Homeostasis. Cell 2020, 183, 110–125. [Google Scholar] [CrossRef]
- Weingartl, H.M.; Sabara, M.; Pasick, J.; van Moorlehem, E.; Babiuk, L. Continuous porcine cell lines developed from alveolar macrophages: Partial characterization and virus susceptibility. J. Virol. Methods 2002, 104, 203–216. [Google Scholar] [CrossRef]
- Joshi, N.; Walter, J.M.; Misharin, A.V. Alveolar Macrophages. Cell Immunol. 2018, 330, 86–90. [Google Scholar] [CrossRef]
- Shabir, N.; Khatun, A.; Nazki, S.; Gu, S.; Lee, S.M.; Hur, T.Y.; Yang, M.S.; Kim, B.; Kim, W.I. In vitro immune responses of porcine alveolar macrophages reflect host immune responses against porcine reproductive and respiratory syndrome viruses. BMC Vet. Res. 2018, 14, 380. [Google Scholar] [CrossRef] [PubMed]
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar] [CrossRef]
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef]
- Mills, R.E.; Walter, K.; Stewart, C.; Handsaker, R.E.; Chen, K.; Alkan, C.; Abyzov, A.; Yoon, S.C.; Ye, K.; Cheetham, R.K.; et al. Mapping copy number variation by population-scale genome sequencing. Nature 2011, 470, 59–65. [Google Scholar] [CrossRef]
- Chaisson, M.J.P.; Sanders, A.D.; Zhao, X.; Malhotra, A.; Porubsky, D.; Rausch, T.; Gardner, E.J.; Rodriguez, O.L.; Guo, L.; Collins, R.L.; et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019, 10, 1784. [Google Scholar] [CrossRef] [PubMed]
- Weischenfeldt, J.; Symmons, O.; Spitz, F.; Korbel, J.O. Phenotypic impact of genomic structural variation: Insights from and for human disease. Nat. Rev. Genet. 2013, 14, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.; Scott, A.J.; Davis, J.R.; Tsang, E.K.; Li, X.; Kim, Y.; Hadzic, T.; Damani, F.N.; Ganel, L.; Consortium, G.T.; et al. The impact of structural variation on human gene expression. Nat. Genet. 2017, 49, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Fang, S.; Wang, J.; Zhang, C.; Zhang, R.; Liu, D.; Zhao, Y.; Hu, X.; Li, N. Genome-wide analysis of structural variants reveals genetic differences in Chinese pigs. PLoS ONE 2017, 12, e0186721. [Google Scholar] [CrossRef]
- Liu, C.; Ran, X.; Wang, J.; Li, S.; Liu, J. Detection of genomic structural variations in Guizhou indigenous pigs and the comparison with other breeds. PLoS ONE 2018, 13, e0194282. [Google Scholar] [CrossRef]
- Zhao, Y.; Hou, Y.; Xu, Y.; Luan, Y.; Zhou, H.; Qi, X.; Hu, M.; Wang, D.; Wang, Z.; Fu, Y.; et al. A compendium and comparative epigenomics analysis of cis-regulatory elements in the pig genome. Nat. Commun. 2021, 12, 2217. [Google Scholar] [CrossRef]
- Zhou, R.; Li, S.T.; Yao, W.Y.; Xie, C.D.; Chen, Z.; Zeng, Z.J.; Wang, D.; Xu, K.; Shen, Z.J.; Mu, Y.; et al. The Meishan pig genome reveals structural variation-mediated gene expression and phenotypic divergence underlying Asian pig domestication. Mol. Ecol. Resour. 2021, 21, 2077–2092. [Google Scholar] [CrossRef]
- Liu, C.; Ran, X.; Yu, C.; Xu, Q.; Niu, X.; Zhao, P.; Wang, J. Whole-genome analysis of structural variations between Xiang pigs with larger litter sizes and those with smaller litter sizes. Genomics 2019, 111, 310–319. [Google Scholar] [CrossRef]
- Gardner, R.C.; Howarth, A.J.; Hahn, P.; Brown-Luedi, M.; Shepherd, R.J.; Messing, J. The complete nucleotide sequence of an infectious clone of cauliflower mosaic virus by M13mp7 shotgun sequencing. Nucleic. Acids. Res. 1981, 9, 2871–2888. [Google Scholar] [CrossRef]
- Vergult, S.; Van Binsbergen, E.; Sante, T.; Nowak, S.; Vanakker, O.; Claes, K.; Poppe, B.; Van der Aa, N.; van Roosmalen, M.J.; Duran, K.; et al. Mate pair sequencing for the detection of chromosomal aberrations in patients with intellectual disability and congenital malformations. Eur. J. Hum. Genet. 2014, 22, 652–659. [Google Scholar] [CrossRef]
- Rowsey, R.A.; Smoley, S.A.; Williamson, C.M.; Vasmatzis, G.; Smadbeck, J.B.; Ning, Y.; Greipp, P.T.; Hoppman, N.L.; Baughn, L.B.; Ketterling, R.P.; et al. Characterization of TCF3 rearrangements in pediatric B-lymphoblastic leukemia/lymphoma by mate-pair sequencing (MPseq) identifies complex genomic rearrangements and a novel TCF3/TEF gene fusion. Blood Cancer J. 2019, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Spielmann, M.; Lupianez, D.G.; Mundlos, S. Structural variation in the 3D genome. Nat. Rev. Genet. 2018, 19, 453–467. [Google Scholar] [CrossRef]
- Stankiewicz, P.; Lupski, J.R. Structural variation in the human genome and its role in disease. Annu. Rev. Med. 2010, 61, 437–455. [Google Scholar] [CrossRef] [PubMed]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.; et al. An integrated map of structural variation in 2,504 human genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef]
- Tubio, J.M.C. Somatic structural variation and cancer. Brief. Funct. Genom. 2015, 14, 339–351. [Google Scholar] [CrossRef][Green Version]
- Macintyre, G.; Ylstra, B.; Brenton, J.D. Sequencing Structural Variants in Cancer for Precision Therapeutics. Trends Genet. 2016, 32, 530–542. [Google Scholar] [CrossRef]
- Li, Y.; Roberts, N.D.; Wala, J.A.; Shapira, O.; Schumacher, S.E.; Kumar, K.; Khurana, E.; Waszak, S.; Korbel, J.O.; Haber, J.E.; et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020, 578, 112–121. [Google Scholar] [CrossRef]
- Ma, H.; Jiang, J.; He, J.; Liu, H.; Han, L.; Gong, Y.; Li, B.; Yu, Z.; Tang, S.; Zhang, Y.; et al. Long-read assembly of the Chinese indigenous Ningxiang pig genome and identification of genetic variations in fat metabolism among different breeds. Mol. Ecol. Resour. 2022, 22, 1508–1520. [Google Scholar] [CrossRef]
- Zheng, X.; Zhao, P.; Yang, K.; Ning, C.; Wang, H.; Zhou, L.; Liu, J. CNV analysis of Meishan pig by next-generation sequencing and effects of AHR gene CNV on pig reproductive traits. J. Anim. Sci. Biotechnol. 2020, 11, 42. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Zhang, X.; Luo, Y.; Liu, R.; Sun, Y.; Zhao, S.; Yu, M.; Cao, J. Large Fragment InDels Reshape Genome Structure of Porcine Alveolar Macrophage 3D4/21 Cells. Genes 2022, 13, 1515. https://doi.org/10.3390/genes13091515
Li X, Zhang X, Luo Y, Liu R, Sun Y, Zhao S, Yu M, Cao J. Large Fragment InDels Reshape Genome Structure of Porcine Alveolar Macrophage 3D4/21 Cells. Genes. 2022; 13(9):1515. https://doi.org/10.3390/genes13091515
Chicago/Turabian StyleLi, Xiaolong, Xiaoqian Zhang, Yandong Luo, Ru Liu, Yan Sun, Shuhong Zhao, Mei Yu, and Jianhua Cao. 2022. "Large Fragment InDels Reshape Genome Structure of Porcine Alveolar Macrophage 3D4/21 Cells" Genes 13, no. 9: 1515. https://doi.org/10.3390/genes13091515
APA StyleLi, X., Zhang, X., Luo, Y., Liu, R., Sun, Y., Zhao, S., Yu, M., & Cao, J. (2022). Large Fragment InDels Reshape Genome Structure of Porcine Alveolar Macrophage 3D4/21 Cells. Genes, 13(9), 1515. https://doi.org/10.3390/genes13091515