
A Chromosome-Length Assembly of the Hawaiian Monk Seal (Neomonachus schauinslandi): A History of “Genetic Purging” and Genomic Stability

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
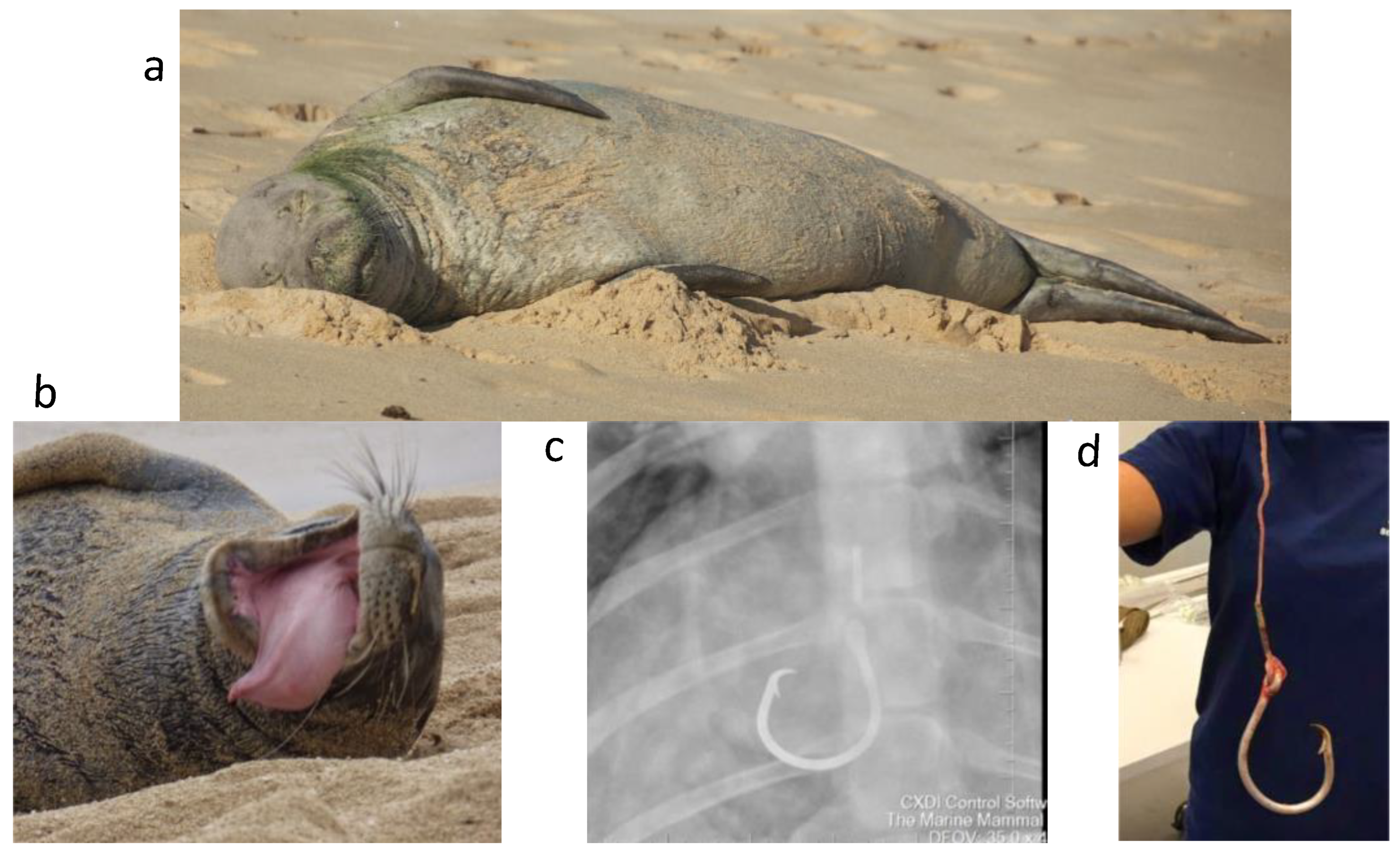
2.1. Sample
2.2. Linked-Read Sequencing
2.3. Nanopore Sequencing
2.4. Bionano Genomics Optical Genome Mapping
2.5. Arima Hi-C and 3D-DNA
2.6. Final Editing
2.7. Haplotype Analysis
2.8. Quality Assessments
2.9. Conserved Synteny Analysis
2.10. Annotation and QC
2.11. Short Read Sequencing and Variant Calling
2.12. Demographic Reconstruction
3. Results
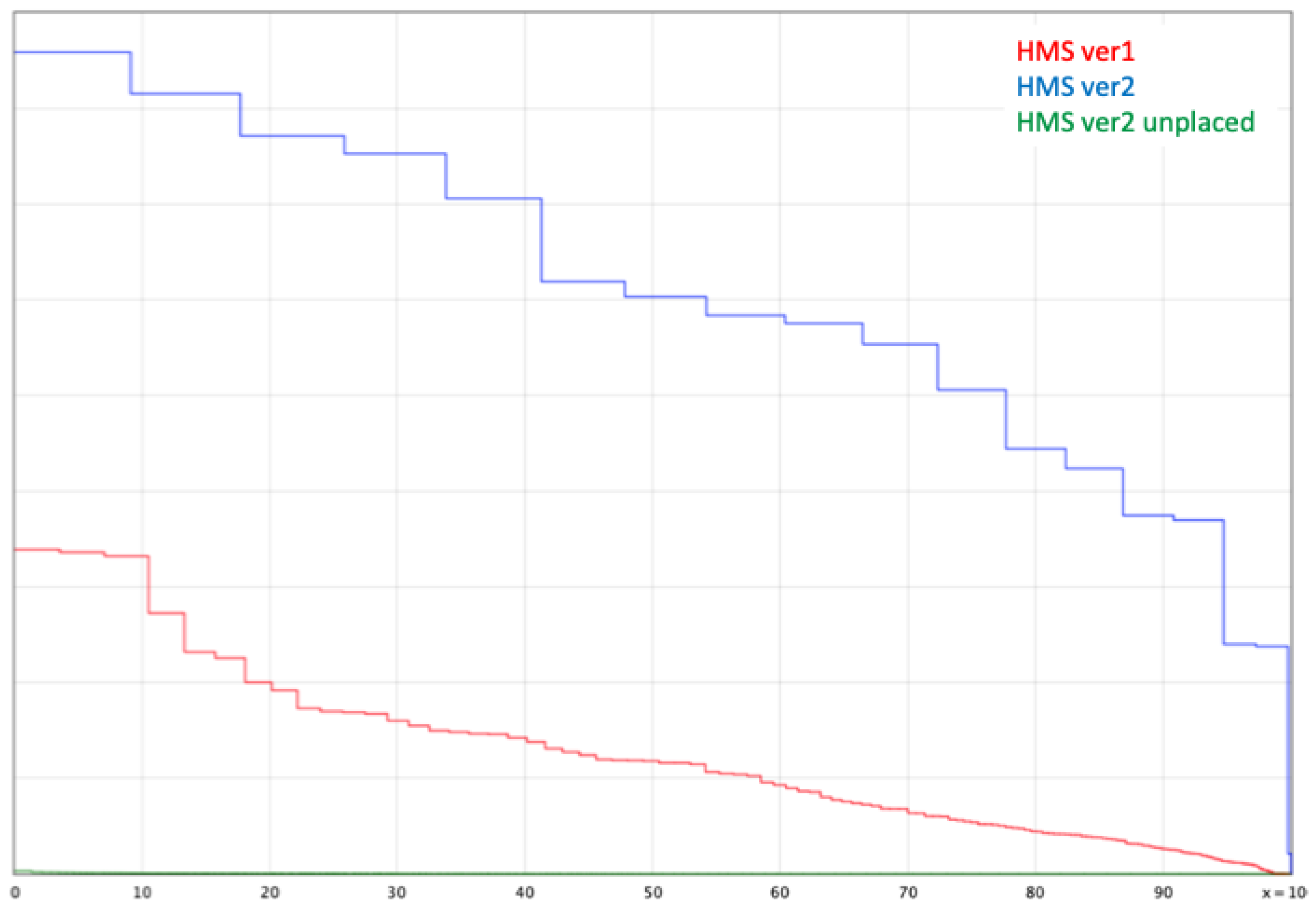
3.1. Genome Assembly and Quality Assessment
3.2. Phylogenic Relationships of “Missing” BUSCO Genes
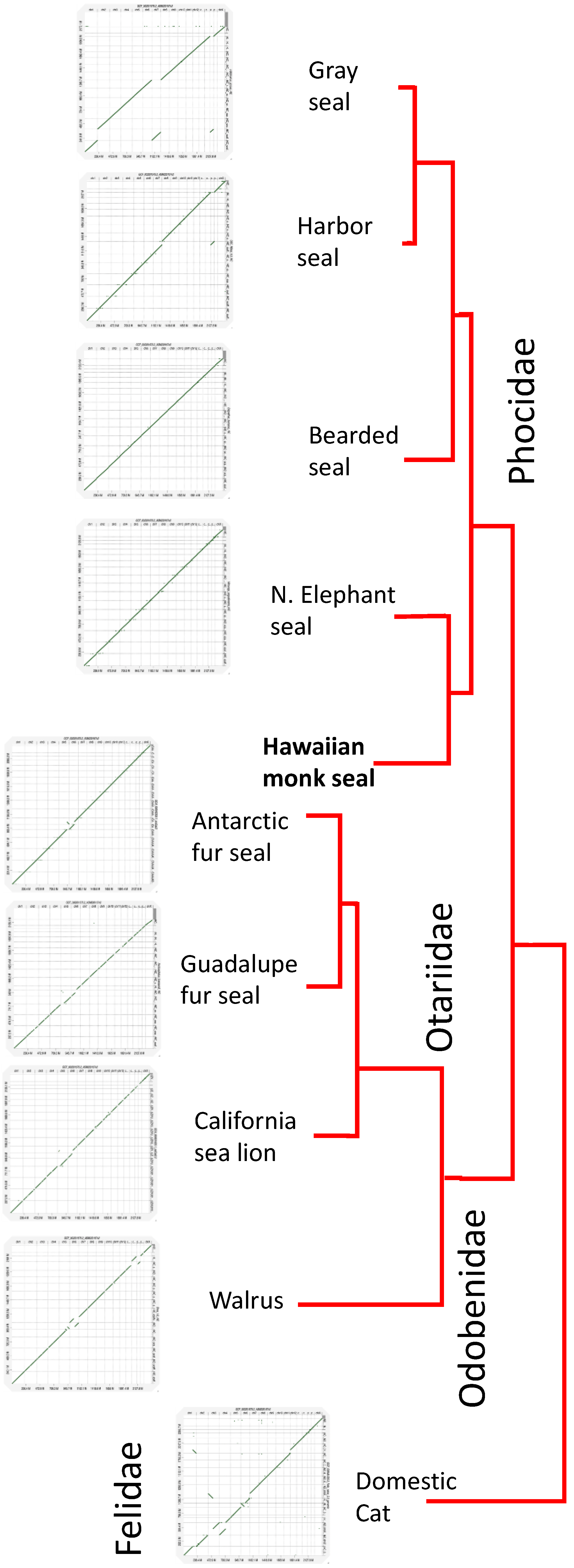
3.3. Conservation of Chromosomal Architecture Based on Genome Alignments
3.4. Heterozygosity Estimates and Phasing
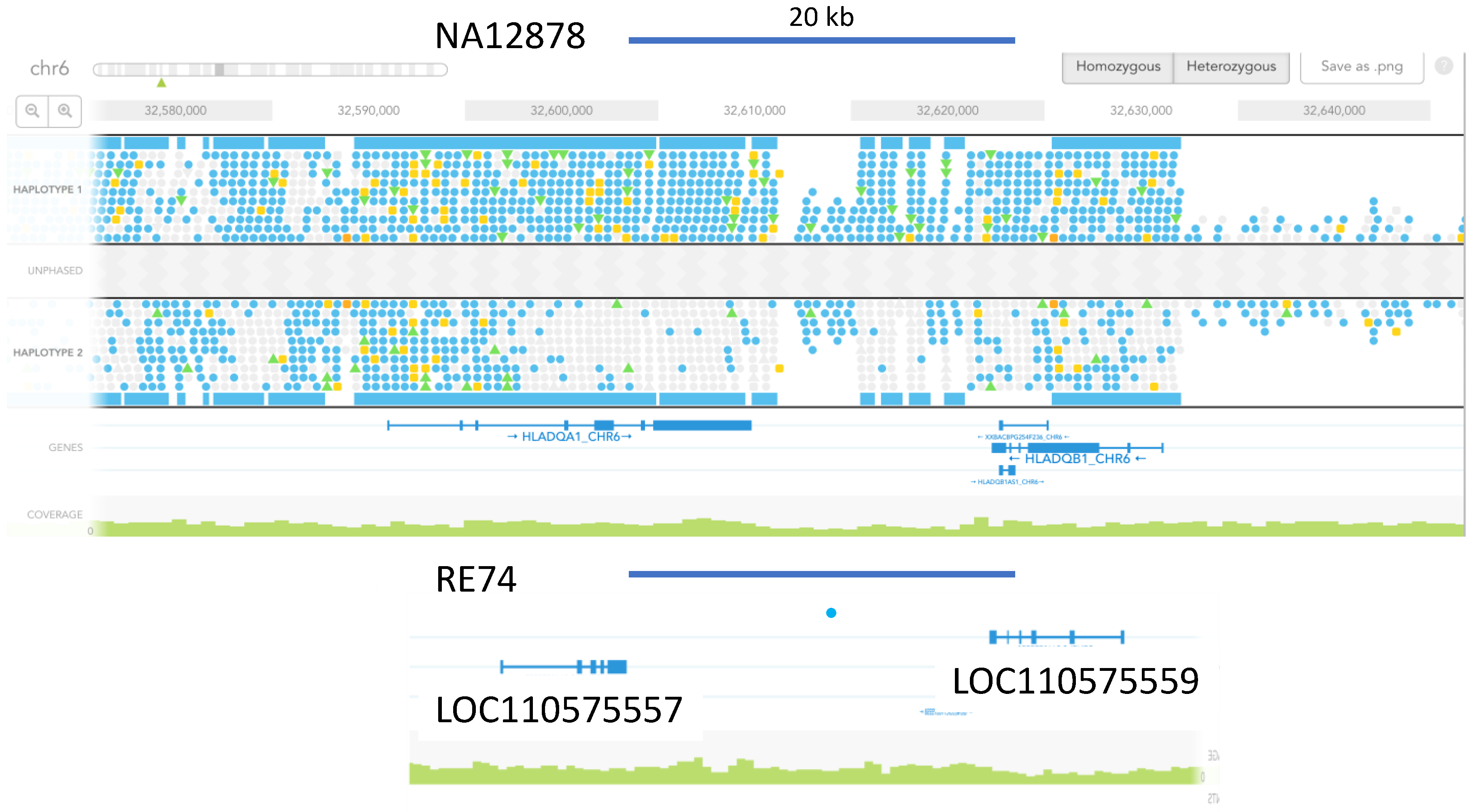
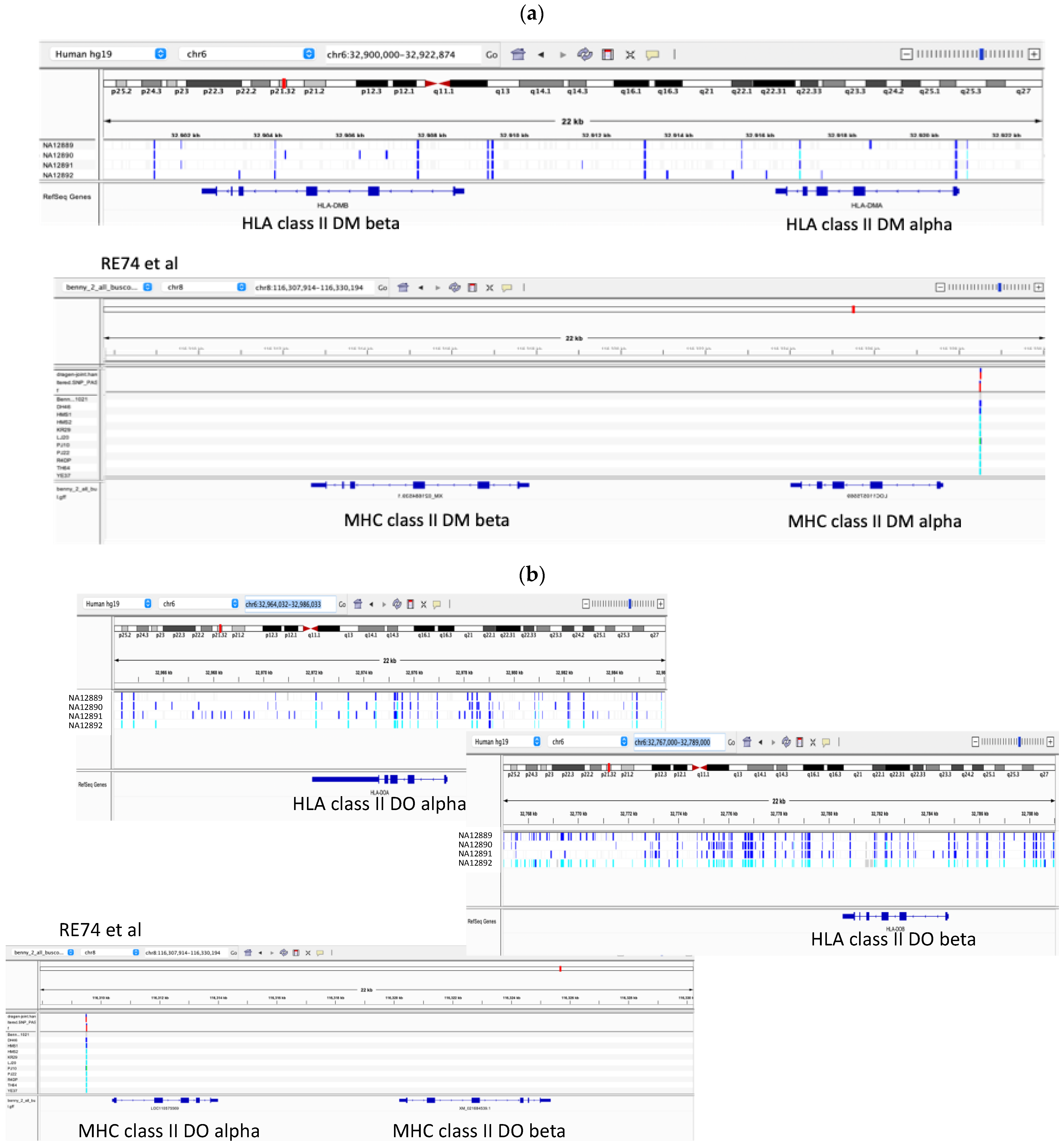
3.5. Marked Loss of MHC Heterogeneity in HMS
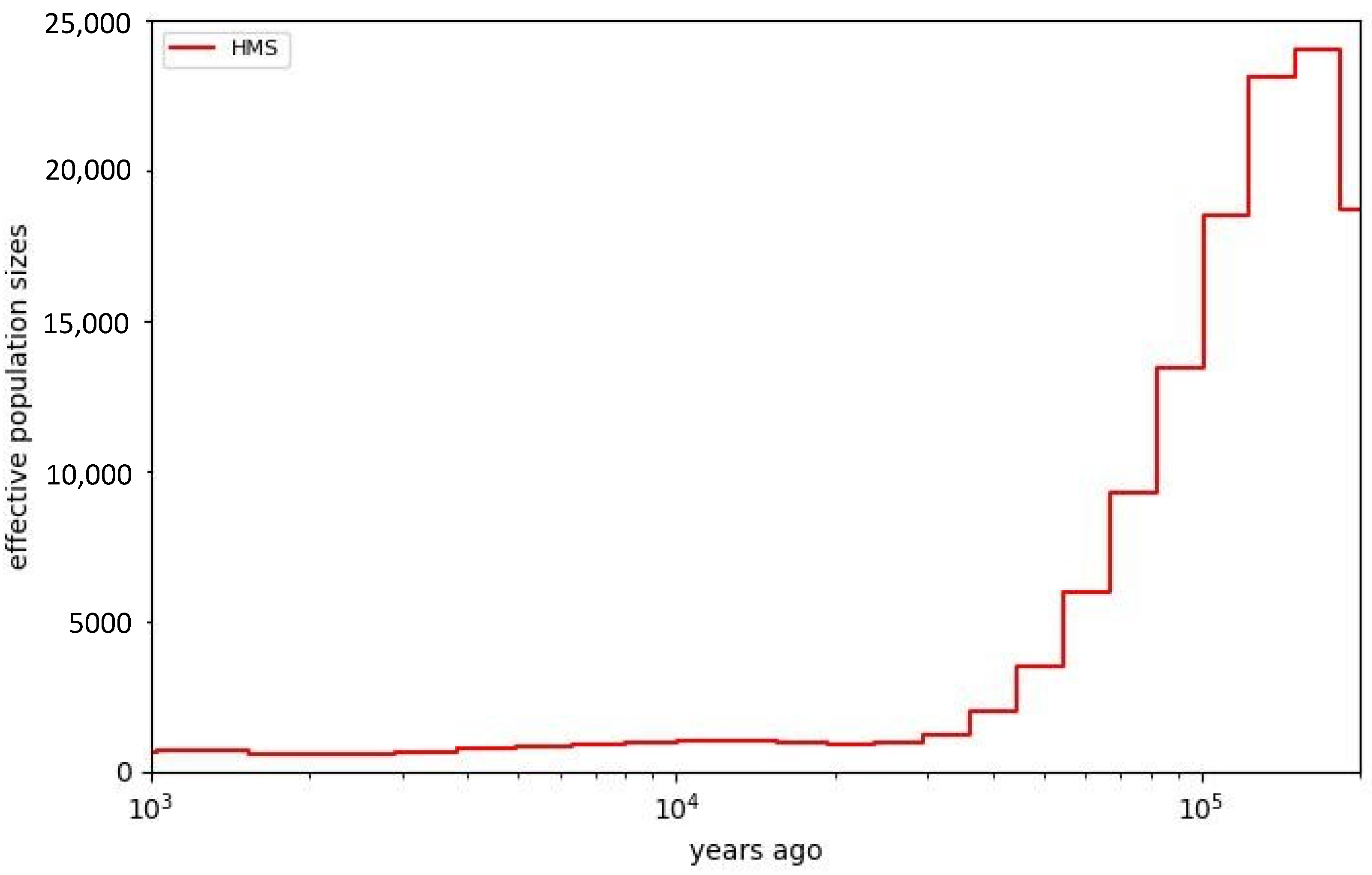
3.6. MSMC Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Slater, G.J.; Scheel, D.-M.; Kolokotronis, S.-O.; Potter, C.W.; Rotstein, D.S.; Tsangaras, K.; Greenwood, A.D.; Helgen, K.M. Biogeography and taxonomy of extinct and endangered monk seals illuminated by ancient DNA and skull morphology. ZooKeys 2014, 409, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Recovery Plan for the Hawaiian Monk Seal Monachus Schauinslandi. Available online: https://repository.library.noaa.gov/view/noaa/3521 (accessed on 6 June 2021).
- Schultz, J.K.; Baker, J.D.; Toonen, R.J.; Bowen, B.W. Extremely Low Genetic Diversity in the Endangered Hawaiian Monk Seal (Monachus schauinslandi). J. Hered. 2009, 100, 25–33. [Google Scholar] [CrossRef]
- Sundaram, L.; Gao, H.; Padigepati, S.R.; McRae, J.F.; Li, Y.; Kosmicki, J.A.; Fritzilas, N.; Hakenberg, J.; Dutta, A.; Shon, J.; et al. Predicting the clinical impact of human mutation with deep neural networks. Nat. Genet. 2018, 50, 1161–1170, Erratum in Nat. Genet. 2019, 51, 364. [Google Scholar] [CrossRef]
- Kim, S.; Cho, Y.S.; Kim, H.-M.; Chung, O.; Kim, H.; Jho, S.; Seomun, H.; Kim, J.; Bang, W.Y.; Kim, C.; et al. Comparison of carnivore, omnivore, and herbivore mammalian genomes with a new leopard assembly. Genome Biol. 2016, 17, 211. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; He, M.; Tang, Q.; Tian, S.; Zhang, J.; Li, Y.; Wang, D.; Jin, L.; Ning, C.; Zhu, W.; et al. Comparative 3D genome architecture in vertebrates. BMC Biol. 2022, 20, 99. [Google Scholar] [CrossRef] [PubMed]
- Duncan, E.J.; Leask, M.P.; Dearden, P.K. Genome Architecture Facilitates Phenotypic Plasticity in the Honeybee (Apis mellifera). Mol. Biol. Evol. 2020, 37, 1964–1978. [Google Scholar] [CrossRef]
- Mohr, D.W.; Naguib, A.; Weisenfeld, N.I.; Kumar, V.; Shah, P.; Church, D.M.; Jaffe, D.; Scott, A.F. Improved de Novo Genome Assembly: Linked-Read Sequencing Combined with Optical Mapping Produce a High Quality Mammalian Genome at Relatively Low Cost. bioRxiv 2017, 128348. [Google Scholar] [CrossRef]
- Weisenfeld, N.I.; Kumar, V.; Shah, P.; Church, D.M.; Jaffe, D.B. Direct determination of diploid genome sequences. Genome Res. 2017, 27, 757–767, Erratum in Genome Res. 2018, 28, 606.1. [Google Scholar] [CrossRef]
- Xiao, M.; Phong, A.; Ha, C.; Chan, T.F.; Cai, D.; Leung, L.; Wan, E.; Kistler, A.L.; DeRisi, J.L.; Selvin, P.R.; et al. Rapid DNA mapping by fluorescent single molecule detection. Nucleic Acids Res. 2007, 35, e16. [Google Scholar] [CrossRef][Green Version]
- Shelton, J.M.; Coleman, M.C.; Herndon, N.; Lu, N.; Lam, E.T.; Anantharaman, T.; Sheth, P.; Brown, S.J. Tools and pipelines for BioNano data: Molecule assembly pipeline and FASTA super scaffolding tool. BMC Genom. 2015, 16, 734. [Google Scholar] [CrossRef]
- Dudchenko, O.; Batra, S.S.; Omer, A.D.; Nyquist, S.K.; Hoeger, M.; Durand, N.C.; Shamim, M.S.; Machol, I.; Lander, E.S.; Aiden, A.P.; et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 2017, 356, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Dudchenko, O.; Shamim, M.S.; Batra, S.S.; Durand, N.C.; Musial, N.T.; Mostofa, R.; Pham, M.; St Hilaire, B.G.; Yao, W.; Stamenova, E.; et al. The Juicebox Assembly Tools module facilitates de novo assembly of mammalian genomes with chromosome-length scaffolds for under $1000. bioRxiv 2018, 254797. [Google Scholar] [CrossRef]
- Robinson, J.T.; Turner, D.; Durand, N.C.; Thorvaldsdóttir, H.; Mesirov, J.P.; Aiden, E.L. Juicebox.js Provides a Cloud-Based Visualization System for Hi-C Data. Cell Syst. 2018, 6, 256–258.e1. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Cabanettes, F.; Klopp, C. D-GENIES: Dot plot large genomes in an interactive, efficient and simple way. PeerJ 2018, 6, e4958. [Google Scholar] [CrossRef]
- Zhao, S.; Agafonov, O.; Azab, A.; Stokowy, T.; Hovig, E. Accuracy and efficiency of germline variant calling pipelines for human genome data. Sci. Rep. 2020, 10, 20222. [Google Scholar] [CrossRef]
- Schiffels, S.; Durbin, R. Inferring human population size and separation history from multiple genome sequences. Nat. Genet. 2014, 46, 919–925. [Google Scholar] [CrossRef]
- Schiffels, S.; Wang, K. MSMC and MSMC2: The Multiple Sequentially Markovian Coalescent. In Statistical Population Genomics; Dutheil, J.Y., Ed.; Methods in Molecular Biology; Humana: New York, NY, USA, 2020; Volume 2090. [Google Scholar] [CrossRef]
- Peart, C.R.; Tusso, S.; Pophaly, S.D.; Botero-Castro, F.; Wu, C.-C.; Aurioles-Gamboa, D.; Baird, A.B.; Bickham, J.W.; Forcada, J.; Galimberti, F.; et al. Author Correction: Determinants of genetic variation across eco-evolutionary scales in pinnipeds. Nat. Ecol. Evol. 2021, 5, 1317, Erratum in Nat. Ecol. Evol. 2020, 4, 1095–1104. [Google Scholar] [CrossRef]
- Schultz, J.K.; Marshall, A.J.; Pfunder, M. Genome-Wide Loss of Diversity in the Critically Endangered Hawaiian Monk Seal. Diversity 2010, 2, 863–880. [Google Scholar] [CrossRef]
- Árnason, Ú. Comparative chromosome studies in Pinnipedia. Hereditas 1974, 76, 179–225. [Google Scholar] [CrossRef] [PubMed]
- Beklemisheva, V.R.; Perelman, P.L.; Lemskaya, N.A.; Proskuryakova, A.A.; Serdyukova, N.A.; Burkanov, V.N.; Gorshunov, M.B.; Ryder, O.; Thompson, M.; Lento, G.; et al. Karyotype Evolution in 10 Pinniped Species: Variability of Heterochromatin versus High Conservatism of Euchromatin as Revealed by Comparative Molecular Cytogenetics. Genes 2020, 11, 1485. [Google Scholar] [CrossRef] [PubMed]
- de Sá, A.L.A.; Breaux, B.; Burlamaqui, T.C.T.; Deiss, T.C.; Sena, L.; Criscitiello, M.F.; Schneider, M.P.C. The Marine Mammal Class II Major Histocompatibility Complex Organization. Front. Immunol. 2019, 10, 696. [Google Scholar] [CrossRef] [PubMed]
- Frazer, J.; Notin, P.; Dias, M.; Gomez, A.; Min, J.K.; Brock, K.; Gal, Y.; Marks, D.S. Disease variant prediction with deep generative models of evolutionary data. Nature 2021, 599, 91–95, Erratum in Nature 2022, 601, E7. [Google Scholar] [CrossRef]
- Prüfer, K.; Racimo, F.; Patterson, N.; Jay, F.; Sankararaman, S.; Sawyer, S.; Heinze, A.; Renaud, G.; Sudmant, P.H.; de Filippo, C.; et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 2014, 505, 43–49. [Google Scholar] [CrossRef]
- Kardos, M.; Armstrong, E.E.; Fitzpatrick, S.W.; Hauser, S.; Hedrick, P.W.; Miller, J.M.; Tallmon, D.A.; Funk, W.C. The crucial role of genome-wide genetic variation in conservation. Proc. Natl. Acad. Sci. USA 2021, 118, e2104642118. [Google Scholar] [CrossRef]
- Robinson, J.A.; Kyriazis, C.C.; Nigenda-Morales, S.F.; Beichman, A.C.; Rojas-Bracho, L.; Robertson, K.M.; Fontaine, M.C.; Wayne, R.K.; Lohmueller, K.E.; Taylor, B.L.; et al. The critically endangered vaquita is not doomed to extinction by inbreeding depression. Science 2022, 376, 635–639. [Google Scholar] [CrossRef]
- Springer, M.S.; Guerrero-Juarez, C.F.; Huelsmann, M.; Collin, M.A.; Danil, K.; McGowen, M.R.; Oh, J.W.; Ramos, R.; Hiller, M.; Plikus, M.V.; et al. Genomic and anatomical comparisons of skin support independent adaptation to life in water by cetaceans and hippos. Curr. Biol. 2021, 31, 2124–2139.e3. [Google Scholar] [CrossRef]
- Becker, S.E.D.; Thomas, R.; Trifonov, V.A.; Wayne, R.K.; Graphodatsky, A.S.; Breen, M. Anchoring the dog to its relatives reveals new evolutionary breakpoints across 11 species of the Canidae and provides new clues for the role of B chromosomes. Chromosom. Res. 2011, 19, 685–708. [Google Scholar] [CrossRef]
- Beklemisheva, V.R.; Perelman, P.L.; Lemskaya, N.A.; Kulemzina, A.L.; Proskuryakova, A.A.; Burkanov, V.N.; Graphodatsky, A.S. The Ancestral Carnivore Karyotype As Substantiated by Comparative Chromosome Painting of Three Pinnipeds, the Walrus, the Steller Sea Lion and the Baikal Seal (Pinnipedia, Carnivora). PLoS ONE 2016, 11, e0147647. [Google Scholar] [CrossRef] [PubMed]
- Nash, W.G.; Menninger, J.C.; Padilla-Nash, H.M.; Stone, G.; Perelman, P.L.; O’Brien, S.J. The Ancestral Carnivore Karyotype (2n = 38) Lives Today in Ringtails. J. Hered. 2008, 99, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Peart, C.R.; Williams, C.; Pophaly, S.D.; Neely, B.A.; Gulland, F.M.D.; Adams, D.J.; Ng, B.L.; Cheng, W.; Goebel, M.E.; Fedrigo, O.; et al. Hi-C scaffolded short- and long-read genome assemblies of the California sea lion are broadly consistent for syntenic inference across 45 million years of evolution. Mol. Ecol. Resour. 2021, 21, 2455–2470. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RE74 | Ma | Pv | Hg | Zc | Or | Clf | Fc | Hs | |
|---|---|---|---|---|---|---|---|---|---|
| Complete | 8845 | 8712 | 8849 | 8542 | 8878 | 8875 | 8742 | 8836 | 8856 |
| Complete & Single-Copy | 8617 | 8467 | 8651 | 8352 | 8543 | 8599 | 8560 | 8761 | 8450 |
| Complete & Duplicated | 228 | 245 | 198 | 190 | 335 | 276 | 182 | 75 | 406 |
| Fragmented | 100 | 175 | 90 | 242 | 83 | 82 | 152 | 111 | 127 |
| Missing | 281 | 339 | 287 | 442 | 265 | 269 | 332 | 279 | 243 |
| Total Missing | HMS RE74 | Ma | Pv | Hg | Zc | Or | Clf | Fc | Hs | |
|---|---|---|---|---|---|---|---|---|---|---|
| TT est. | - | 13.5 | 18.4 | 18.4 | 26 | 26 | 46 | 54 | 96 | |
| 281 | HMS | - | 188 | 185 | 179 | 164 | 167 | 129 | 128 | 99 |
| 339 | Ma | - | 186 | 185 | 153 | 155 | 127 | 133 | 100 | |
| 332 | Pv | - | 207 | 172 | 170 | 131 | 135 | 111 | ||
| 332 | Hg | - | 160 | 166 | 125 | 133 | 108 | |||
| 265 | Zc | - | 184 | 133 | 126 | 106 | ||||
| 269 | Or | - | 127 | 134 | 103 | |||||
| 332 | Clf | - | 122 | 104 | ||||||
| 279 | Fc | - | 106 | |||||||
| 243 | Hs | - |
| Position/Mb | Locus ID | RNA Sequence | NCBI Annotation |
|---|---|---|---|
| 2.66 | LOC110582119 | XM_021692074.1 | ubiquitin-like modifier-activating enzyme 1 |
| 2.64 | LOC110582047 | XM_021691967.1 | BCL-6 corepressor-like |
| 2.89 | LOC110581036 | XM_021690741.1 | γ-taxilin-like |
| 2.95 | SRY | XM_021693078.1 | sex-determining region Y, SRY |
| 3.05 | LOC110582200 | XM_021692141.1 | cullin-4B-like |
| 3.33 | LOC110591777 | XM_021702682.1 | eukaryotic translation initiation factor 1A, X-chromosomal |
| 3.42 | LOC110591779 | XM_021702683.1 | heat shock transcription factor, Y-linked-like |
| 3.50 | LOC110591787 | XM_021702687.1 | lysine-specific demethylase 5D |
| 3.56 | LOC110591776 | XM_021702680.1 | zinc finger X-chromosomal protein-like |
| 3.66 | LOC110591775 | XM_021702679.1 | eukaryotic translation initiation factor 2 subunit 3, Y-linked-like |
| 4.41 | OFD1 | XM_021692108.1 | OFD1, centriole and centriolar satellite protein (OFD1), partial mRNA |
| 4.46 | LOC110580574 | XM_021690271.1 | histone demethylase UTY-like |
| 4.66 | LOC110581163 | XM_021690923.1 | ATP-dependent RNA helicase DDX3X-like |
| 4.70 | LOC110581157 | XM_021690911.1 | probable ubiquitin carboxyl-terminal hydrolase FAF-X |
| 4.81 | LOC110580811 | XM_021690464.1 | RNA-binding motif protein, X chromosome-like |
| 4.83 | LOC110580801 | XM_021690458.1 | probable ubiquitin carboxyl-terminal hydrolase FAF-Y |
| 4.89 | LOC110580822 | XM_021690474.1 | serine protease 55-like |
| 4.91 | LOC110580830 | XM_021690484.1 | serine protease 52-like |
| 5.17 | LOC110581101 | XM_021690826.1 | thymosin β-4-like |
| 5.25 | LOC110584133 | XM_021694172.1 | ras-related protein Rab-9A-like |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohr, D.W.; Gaughran, S.J.; Paschall, J.; Naguib, A.; Pang, A.W.C.; Dudchenko, O.; Aiden, E.L.; Church, D.M.; Scott, A.F. A Chromosome-Length Assembly of the Hawaiian Monk Seal (Neomonachus schauinslandi): A History of “Genetic Purging” and Genomic Stability. Genes 2022, 13, 1270. https://doi.org/10.3390/genes13071270
Mohr DW, Gaughran SJ, Paschall J, Naguib A, Pang AWC, Dudchenko O, Aiden EL, Church DM, Scott AF. A Chromosome-Length Assembly of the Hawaiian Monk Seal (Neomonachus schauinslandi): A History of “Genetic Purging” and Genomic Stability. Genes. 2022; 13(7):1270. https://doi.org/10.3390/genes13071270
Chicago/Turabian StyleMohr, David W., Stephen J. Gaughran, Justin Paschall, Ahmed Naguib, Andy Wing Chun Pang, Olga Dudchenko, Erez Lieberman Aiden, Deanna M. Church, and Alan F. Scott. 2022. "A Chromosome-Length Assembly of the Hawaiian Monk Seal (Neomonachus schauinslandi): A History of “Genetic Purging” and Genomic Stability" Genes 13, no. 7: 1270. https://doi.org/10.3390/genes13071270
APA StyleMohr, D. W., Gaughran, S. J., Paschall, J., Naguib, A., Pang, A. W. C., Dudchenko, O., Aiden, E. L., Church, D. M., & Scott, A. F. (2022). A Chromosome-Length Assembly of the Hawaiian Monk Seal (Neomonachus schauinslandi): A History of “Genetic Purging” and Genomic Stability. Genes, 13(7), 1270. https://doi.org/10.3390/genes13071270