
MicroRNA Processing Pathway-Based Polygenic Score for Clear Cell Renal Cell Carcinoma in the Volga-Ural Region Populations of Eurasian Continent

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Sample
2.2. Blood Sample Collection, DNA Extraction, and Genotyping Procedures
2.3. Quality Control (QC) and Association Analysis
2.4. PGS Calculation
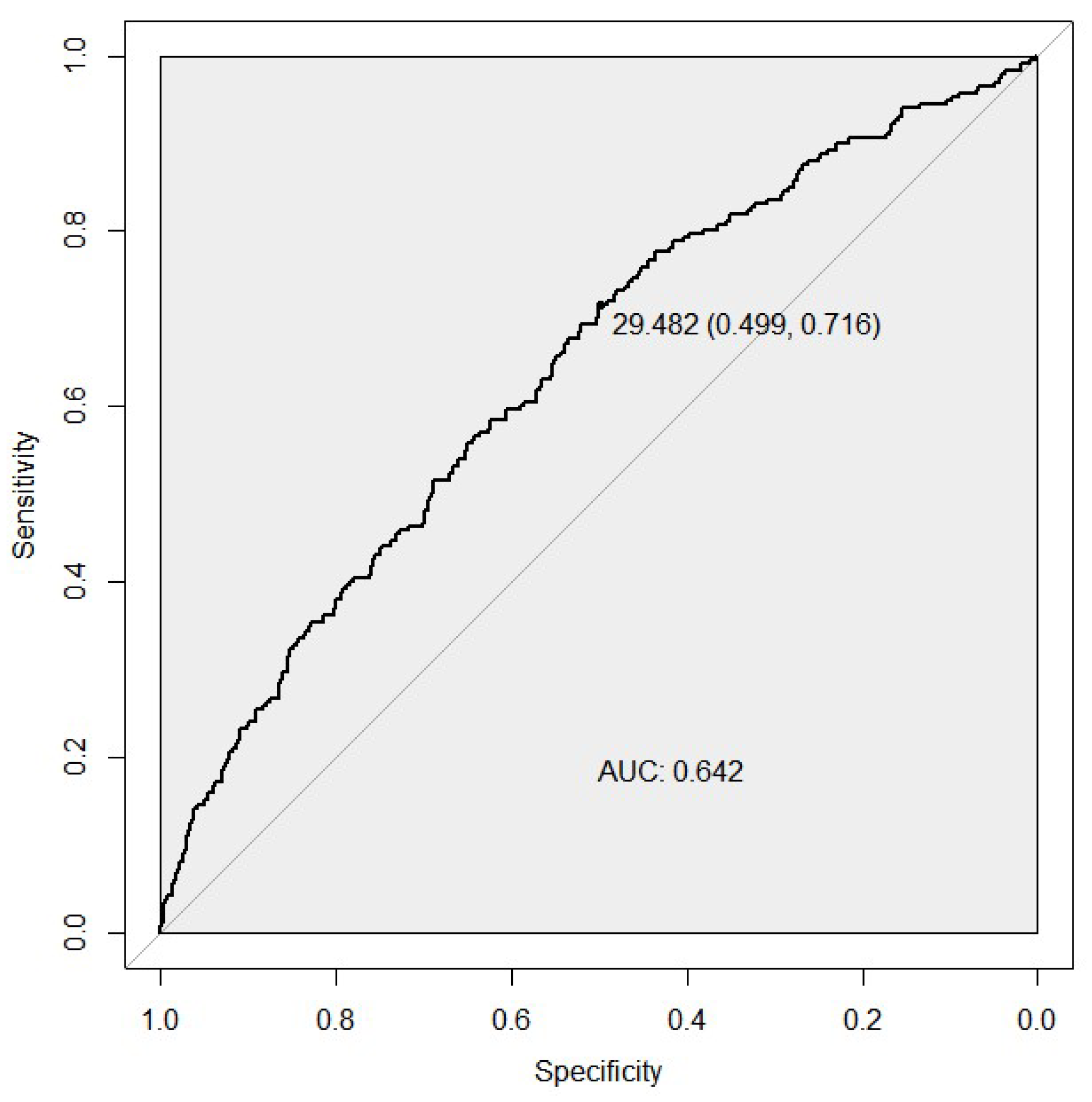
2.5. Receiver Operating Characteristic (ROC) Analysis
3. Results
3.1. Association Analysis

{kind=link}
| Gene Name | Chromosome | Position, (GRCh37) | rsID | Risk Allele/Non-Risk Allele | Effect Allele Frequency Cases/Controls | OR (95% CI) | p-Value |
|---|---|---|---|---|---|---|---|
| AGO1 | 1 | 36,380,133 | rs595055 | T/C | 0.26/0.30 | 1.20 (0.71–0.99) | 0.04 |
| DDX20 | 1 | 112,308,953 | rs197412 | T/C | 0.47/0.49 | 1.11 (0.77–1.06) | 0.21 |
| DROSHA | 5 | 31,435,627 | rs4867329 | A/C | 0.46/0.49 | 1.11 (0.77–1.05) | 0.19 |
| C5orf22 | 5 | 31,532,789 | rs17409893 | A/G | 0.31/0.32 | 1.08 (0.78–1.10) | 0.37 |
| XPO5 | 6 | 43,492,578 | rs2257082 | G/A | 0.33/0.33 | 1.00 (0.84–1.18) | 0.98 |
| AGO2 | 8 | 141,555,862 | rs3864659 | A/C | 0.12/0.14 | 1.15 (0.69–1.10) | 0.24 |
| AGO2 | 8 | 141,594,460 | rs7005286 | T/C | 0.23/0.21 | 1.11 (0.93–1.33) | 0.26 |
| MIR196A2 | 12 | 54,385,599 | rs11614913 | T/C | 0.38/0.37 | 1.06 (0.91–1.25) | 0.45 |
| PIWIL1 | 12 | 130,852,174 | rs11060845 | G/T | 0.07/0.11 | 1.69 (0.44–0.79) | 4.32 × 10−4 |
| RAN | 12 | 131,355,546 | rs3809142 | C/T | 0.11/0.16 | 1.57 (0.50–0.81) | 3.05 × 10−4 |
| DICER1 | 14 | 95,554,142 | rs1057035 | C/T | 0.44/0.29 | 1.85 (1.58-2.18) | 4.05 × 10−14 |
| DICER1 | 14 | 95,556,747 | rs13078 | T/A | 0.13/0.17 | 1.28 (0.62–0.98) | 0.03 |
| GEMIN4 | 17 | 649,232 | rs3744741 | C/T | 0.15/0.18 | 1.19 (0.68–1.04) | 0.11 |
| GEMIN4 | 17 | 649,505 | rs4968104 | T/A | 0.21/0.22 | 1.06 (0.78–1.14) | 0.55 |
| GEMIN4 | 17 | 649,935 | rs2740348 | G/C | 0.19/0.17 | 1.20 (0.98–1.47) | 0.07 |
| microRNA-423 (NSRP1) | 17 | 28,444,183 | rs6505162 | A/C | 0.50/0.46 | 1.17 (1.00–1.37) | 0.04 |
| DDX5 | 17 | 62,502,435 | rs1991401 | G/A | 0.44/0.38 | 1.25 (1.07–1.47) | 4.99 × 10−3 |
| MIR27A | 19 | 13,947,292 | rs895819 | T/C | 0.34/0.35 | 1.06 (0.80–1.11) | 0.45 |
| DGCR8 | 22 | 20,098,544 | rs417309 | G/A | 0.08/0.10 | 1.17 (0.65–1.12) | 0.26 |
| DGCR8 | 22 | 20,098,582 | rs720012 | A/G | 0.24/0.20 | 1.24 (1.03–1.49) | 0.02 |
| DGCR8 | 22 | 20,098,882 | rs720014 | C/T | 0.24/0.21 | 1.15 (0.95–1.40) | 0.14 |
3.2. Receiver Operator Characteristic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Singh, D. Current updates and future perspectives on the management of renal cell carcinoma. Life Sci. 2020, 264, 118632. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Xu, F.; Tian, J.; Wang, Y.; Guo, N.; Wan, Z.; He, M.; Gao, M.; Gao, K.; Chong, T. Prognostic value of circulating tumor cells and immune-inflammatory cells in patients with renal cell carcinoma. Urol. Oncol. 2022, 40, e21–e167. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, France, 2020; Available online: https://gco.iarc.fr/today (accessed on 22 March 2022).
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Prim. 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Maher, E.R. Hereditary renal cell carcinoma syndromes: Diagnosis, surveillance and management. World J. Urol. 2018, 36, 1891–1898. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, E.; Demidova, E.V.; Iqbal, W.; Serebriiskii, I.G.; Vlasenkova, R.; Ghatalia, P.; Zhou, Y.; Rainey, K.; Forman, A.F.; Dunbrack, R.L.; et al. Interaction of germline variants in a family with a history of early-onset clear cell renal cell carcinoma. Mol. Genet. Genomic Med. 2019, 7, e556. [Google Scholar] [CrossRef]
- Wong, E.C.L.; Breau, R.H.; Mallick, R.; Wood, L.; Pouliot, F.; Basappa, N.S.; Tanguay, S.; Soulières, D.; So, A.; Heng, D.; et al. Renal Cell Carcinoma in the Canadian Indigenous Population. Curr. Oncol. 2019, 26, 367–371. [Google Scholar] [CrossRef] [Green Version]
- Mavaddat, N.; Pharoah, P.D.P.; Michailidou, K.; Tyrer, J.; Brook, M.; Bolla, M.K.; Wang, Q.; Dennis, J.; Dunning, A.M.; Shah, M.; et al. Prediction of Breast Cancer Risk Based on Profiling with Common Genetic Variants. J. Natl. Cancer Inst. 2015, 107, djv036. [Google Scholar] [CrossRef]
- Gao, B.; Wang, L.; Zhang, Y.; Zhang, N.; Han, M.; Liu, H.; Sun, D.; Liu, Y. MiR-532-3p suppresses cell viability, migration and invasion of clear cell renal cell carcinoma through targeting TROAP. Cell Cycle 2021, 20, 1578–1588. [Google Scholar] [CrossRef]
- Kalantzakos, T.J.; Sullivan, T.B.; Gloria, T.; Canes, D.; Moinzadeh, A.; Rieger-Christ, K.M. MiRNA-424-5p Suppresses Proliferation, Migration, and Invasion of Clear Cell Renal Cell Carcinoma and Attenuates Expression of O-GlcNAc-Transferase. Cancers 2021, 13, 5160. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, J.; Hu, X.; Ge, Q.; Xiao, J.; Ginting, C.N. Expression of miR-410 in peripheral blood of patients with clear cell renal cell carcinoma and its effect on proliferation and invasion of Caki-2 cells. J. BUON 2021, 26, 2059–2066. Available online: https://pubmed.ncbi.nlm.nih.gov/34761617/ (accessed on 3 June 2022).
- Shi, L.; Wang, M.; Li, H.; You, P. MicroRNAs in Body Fluids: A More Promising Biomarker for Clear Cell Renal Cell Carcinoma. Cancer Manag. Res. 2021, 13, 7663–7675. [Google Scholar] [CrossRef] [PubMed]
- Chalbatani, G.M.; Momeni, S.A.; Hadloo, M.H.M.; Karimi, Z.; Hadizadeh, M.; Jalali, S.A.; Miri, S.R.; Memari, F.; Hamblin, M.R. Comprehensive analysis of ceRNA networks to determine genes related to prognosis, overall survival, and immune infiltration in clear cell renal carcinoma. Comput. Biol. Med. 2021, 141, 105043. [Google Scholar] [CrossRef]
- Lee, S.S.; Min, H.; Ha, J.Y.; Kim, B.H.; Choi, M.S.; Kim, S. Dysregulation of the miRNA biogenesis components DICER1, DROSHA, DGCR8 and AGO2 in clear cell renal cell carcinoma in both a Korean cohort and the cancer genome atlas kidney clear cell carcinoma cohort. Oncol. Lett. 2019, 18, 4337–4345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, B.M.; Robles, A.I.; Harris, C.C. Genetic variation in microRNA networks: The implications for cancer research. Nat. Rev. Cancer 2010, 10, 389–402. [Google Scholar] [CrossRef]
- Fabbri, M.; Valeri, N.; Calin, G.A. MicroRNAs and genomic variations: From Proteus tricks to Prometheus gift. Carcinogenesis 2009, 30, 912–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Cordoba, S.L.; Salido-Guadarrama, I.; Rodriguez-Dorantes, M.; Hidalgo-Miranda, A. miRNA biogenesis: Biological impact in the development of cancer. Cancer Biol. Ther. 2014, 15, 1444–1455. [Google Scholar] [CrossRef] [Green Version]
- Hata, A.; Kashima, R. Dysregulation of microRNA biogenesis machinery in cancer. Crit. Rev. Biochem. Mol. Biol. 2015, 51, 121–134. [Google Scholar] [CrossRef]
- World Medical Association. World Medical Association Declaration of Helsinki: Ethical principles for medical research involving human subjects. JAMA 2013, 310, 2191–2194. [Google Scholar] [CrossRef] [Green Version]
- Sherry, S.T.; Ward, M.; Sirotkin, K. dbSNP—Database for Single Nucleotide Polymorphisms and Other Classes of Minor Genetic Variation. Genome Res. 1999, 9, 677–679. [Google Scholar] [CrossRef]
- Gibbs, R.A.; Belmont, J.W.; Hardenbol, P.; Willis, T.D.; Yu, F.L.; Yang, H.M.; Ch’ang, L.Y.; Huang, W.; Liu, B.; Shen, Y.; et al. The international HapMap project. Nature 2003, 426, 789–796. [Google Scholar] [CrossRef] [Green Version]
- Griffiths-Jones, S.; Grocock, R.J.; Van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022; Available online: www.R-project.org (accessed on 22 March 2022).
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Baker, E.; Schmidt, K.M.; Sims, R.; O’Donovan, M.C.; Williams, J.; Holmans, P.; Escott-Price, V.; GERAD Consortium. POLARIS: Polygenic LD-adjusted risk score approach for set-based analysis of GWAS data. Genet. Epidemiol. 2018, 42, 366–377. [Google Scholar] [CrossRef] [Green Version]
- Mucci, L.A.; Hjelmborg, J.B.; Harris, J.R.; Czene, K.; Havelick, D.J.; Scheike, T.; Graff, R.E.; Holst, K.; Möller, S.; Unger, R.H.; et al. Familial Risk and Heritability of Cancer among Twins in Nordic Countries. JAMA J. Am. Med. Assoc. 2016, 315, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Haas, N.B.; Nathanson, K.L. Hereditary Kidney Cancer Syndromes. Adv. Chronic Kidney Dis. 2014, 21, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Scelo, G.; Purdue, M.P.; Brown, K.M.; Johansson, M.; Wang, Z.; Eckel-Passow, J.E.; Ye, Y.; Hofmann, J.; Choi, J.; Foll, M.; et al. Genome-wide association study identifies multiple risk loci for renal cell carcinoma. Nat. Commun. 2017, 8, 15724. [Google Scholar] [CrossRef]
- Guo, Z.; Shu, Y.; Zhou, H.; Zhang, W. Identification of diagnostic and prognostic biomarkers for cancer: Focusing on genetic variations in microRNA regulatory pathways. Mol. Med. Rep. 2016, 13, 1943–1952. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.J.; Mishra, P.J.; Banerjee, D.; Bertino, J.R. MiRSNPs or MiR-polymorphisms, new players in microRNA mediated regulation of the cell: Introducing microRNA pharmacogenomics. Cell Cycle 2008, 7, 853–858. [Google Scholar] [CrossRef] [Green Version]
- Horikawa, Y.; Wood, C.G.; Yang, H.; Zhao, H.; Ye, Y.; Gu, J.; Lin, J.; Habuchi, T.; Wu, X. Single Nucleotide Polymorphisms of microRNA Machinery Genes Modify the Risk of Renal Cell Carcinoma. Clin. Cancer Res. 2008, 14, 7956–7962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purdue, M.P.; Rhee, J.; Moore, L.; Gao, X.; Sun, X.; Kirk, E.; Bencko, V.; Janout, V.; Mates, D.; Zaridze, D.; et al. Differences in risk factors for molecular subtypes of clear cell renal cell carcinoma. Int. J. Cancer 2021, 149, 1448–1454. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Tsai, C.-W.; Chang, W.-S.; Xu, J.; Xu, Y.; Bau, D.-T.; Gu, J. Prognostic value of leukocyte telomere length in renal cell carcinoma patients. Am. J. Cancer Res. 2020, 10, 3428–3439. Available online: https://pubmed.ncbi.nlm.nih.gov/33163281/ (accessed on 3 June 2022). [PubMed]
- Jia, G.; Lu, Y.; Wen, W.; Long, J.; Liu, Y.; Tao, R.; Li, B.; Denny, J.C.; Shu, X.-O.; Zheng, W. Evaluating the Utility of Polygenic Risk Scores in Identifying High-Risk Individuals for Eight Common Cancers. JNCI Cancer Spectr. 2020, 4, pkaa021. [Google Scholar] [CrossRef]

| Characteristic | Individuals with ccRCC | Controls |
|---|---|---|
| Total | 464 | 1042 |
| Sex, n (%) | ||
| Male | 282 (60.8) | 519 (49.8) |
| Female | 182 (39.2) | 523 (50.2) |
| Age, years, mean ± SD | 56.01±0.71 | 53.6 ± 0.66 |
| TNM stage | ||
| I-II, n (%) | 267 (57.5) | – |
| III-IV, n (%) | 197 (42.5) | – |
| Ethnicity, n (%) | ||
| Bashkir | 78 (16.8) | 142 (13.6) |
| Tatar | 174 (37.5) | 457 (43.9) |
| Russian | 212 (45.7) | 443 (42.5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanova, E.; Gilyazova, I.; Pavlov, V.; Izmailov, A.; Gimalova, G.; Karunas, A.; Prokopenko, I.; Khusnutdinova, E. MicroRNA Processing Pathway-Based Polygenic Score for Clear Cell Renal Cell Carcinoma in the Volga-Ural Region Populations of Eurasian Continent. Genes 2022, 13, 1281. https://doi.org/10.3390/genes13071281
Ivanova E, Gilyazova I, Pavlov V, Izmailov A, Gimalova G, Karunas A, Prokopenko I, Khusnutdinova E. MicroRNA Processing Pathway-Based Polygenic Score for Clear Cell Renal Cell Carcinoma in the Volga-Ural Region Populations of Eurasian Continent. Genes. 2022; 13(7):1281. https://doi.org/10.3390/genes13071281
Chicago/Turabian StyleIvanova, Elizaveta, Irina Gilyazova, Valentin Pavlov, Adel Izmailov, Galiya Gimalova, Alexandra Karunas, Inga Prokopenko, and Elza Khusnutdinova. 2022. "MicroRNA Processing Pathway-Based Polygenic Score for Clear Cell Renal Cell Carcinoma in the Volga-Ural Region Populations of Eurasian Continent" Genes 13, no. 7: 1281. https://doi.org/10.3390/genes13071281
APA StyleIvanova, E., Gilyazova, I., Pavlov, V., Izmailov, A., Gimalova, G., Karunas, A., Prokopenko, I., & Khusnutdinova, E. (2022). MicroRNA Processing Pathway-Based Polygenic Score for Clear Cell Renal Cell Carcinoma in the Volga-Ural Region Populations of Eurasian Continent. Genes, 13(7), 1281. https://doi.org/10.3390/genes13071281