
Estimations of Mutation Rates Depend on Population Allele Frequency Distribution: The Case of Autosomal Microsatellites

 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
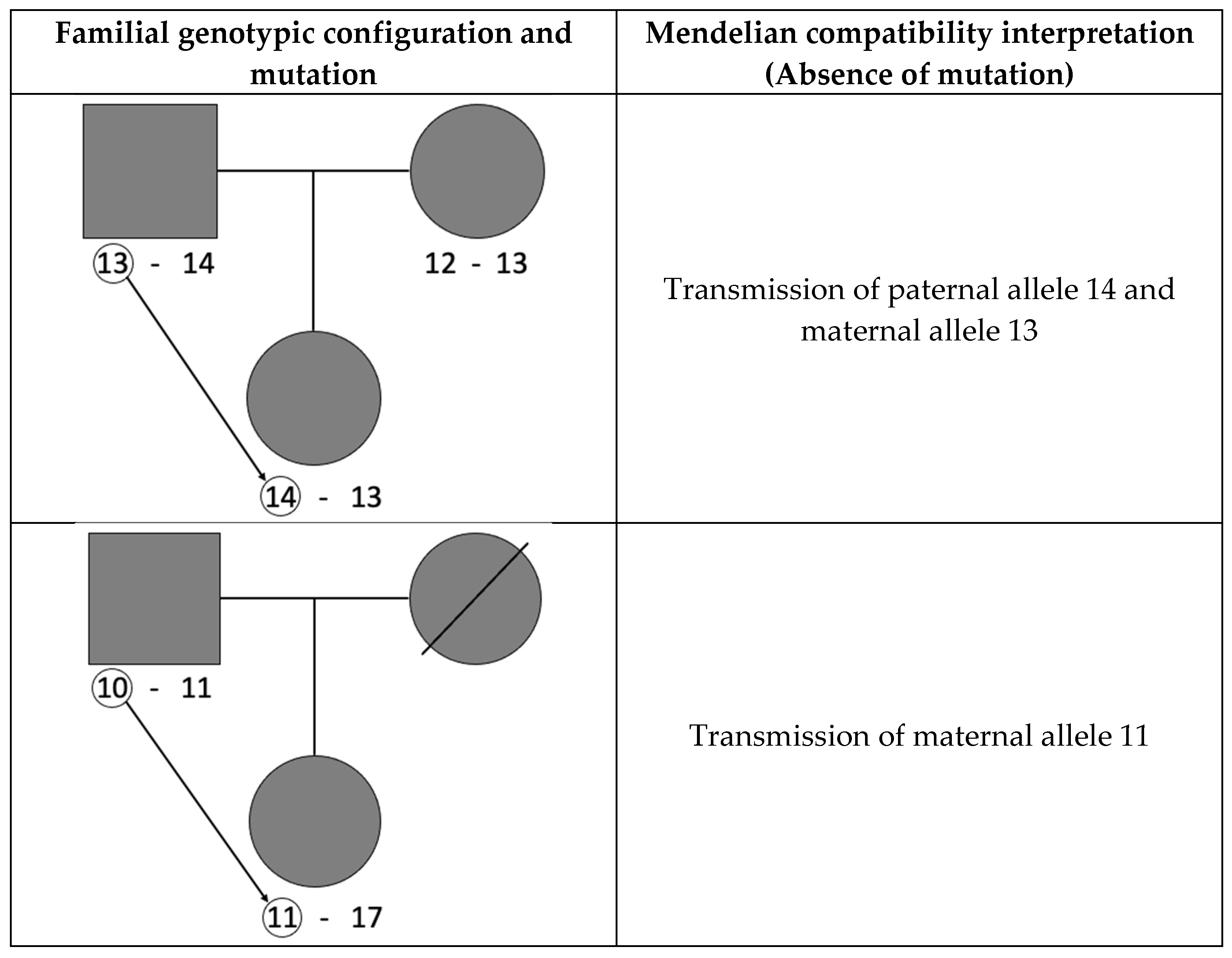
2. Materials and Methods
3. Results
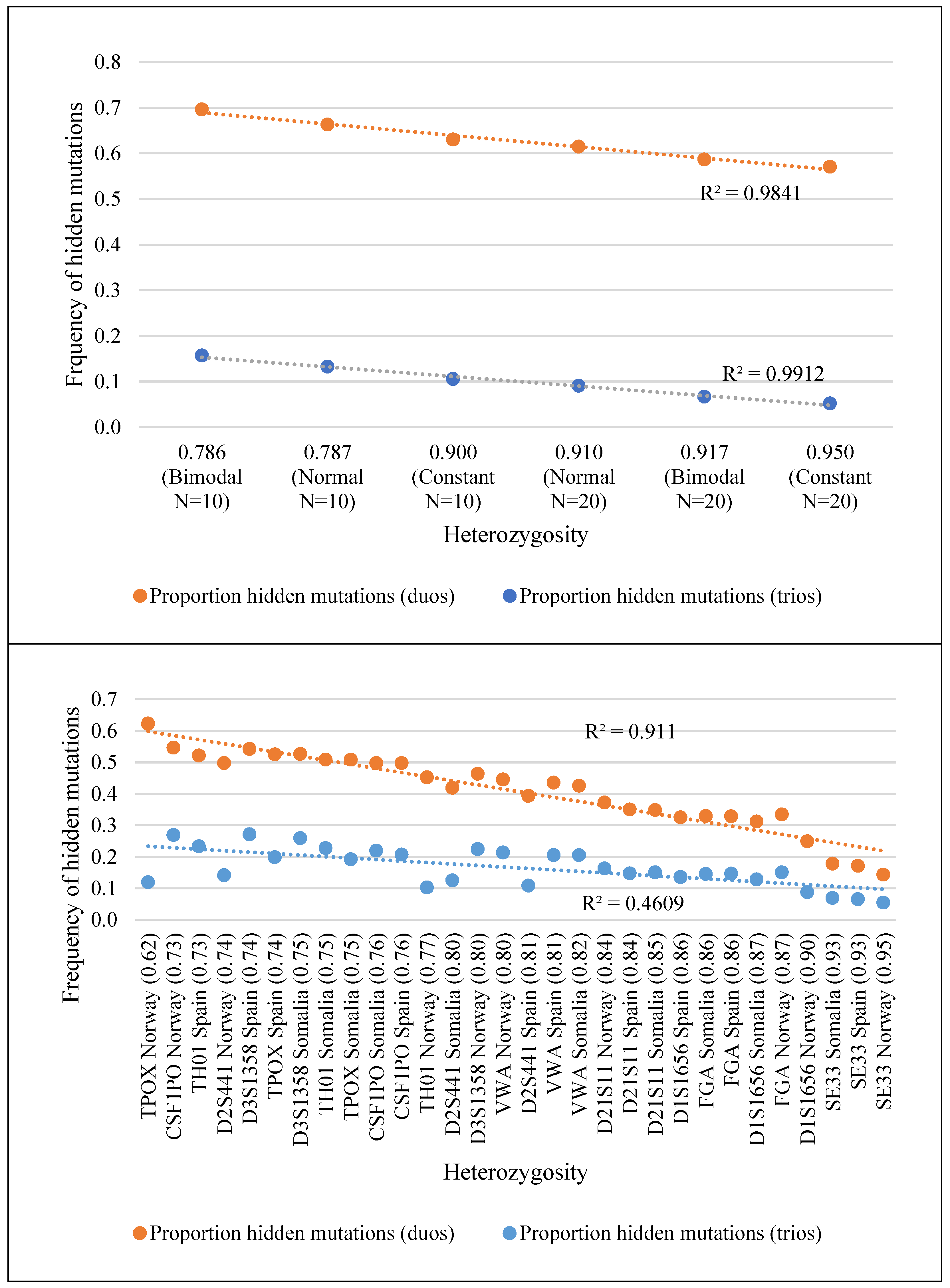
3.1. Real Allele Frequency Distributions
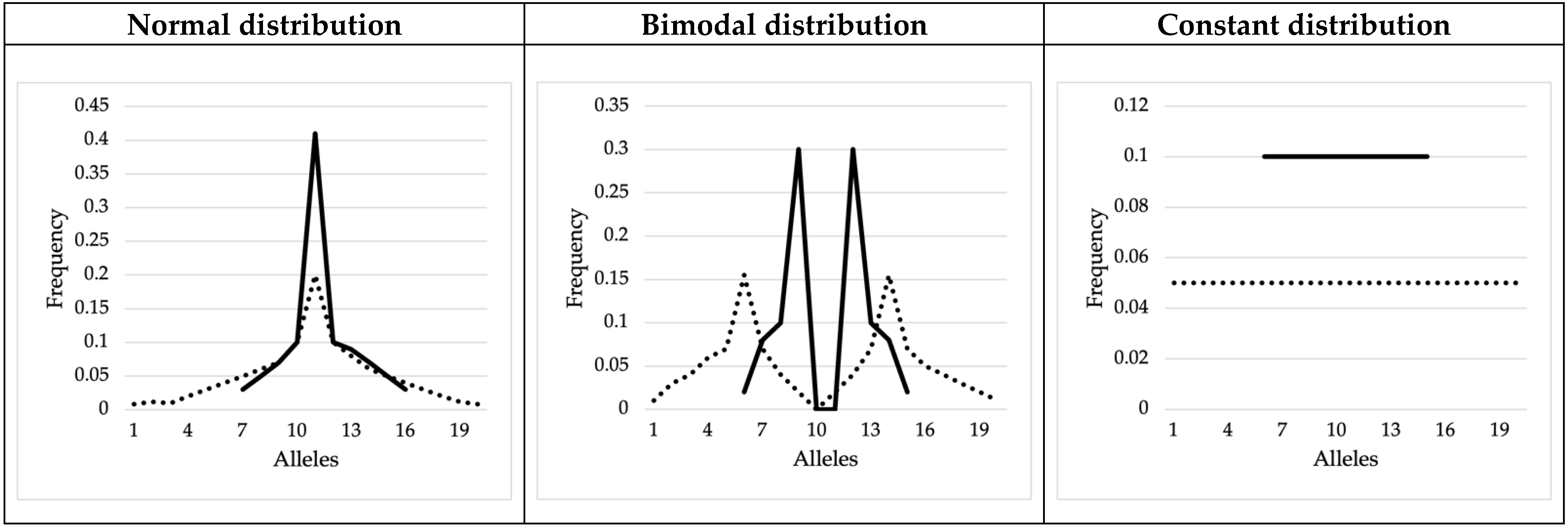
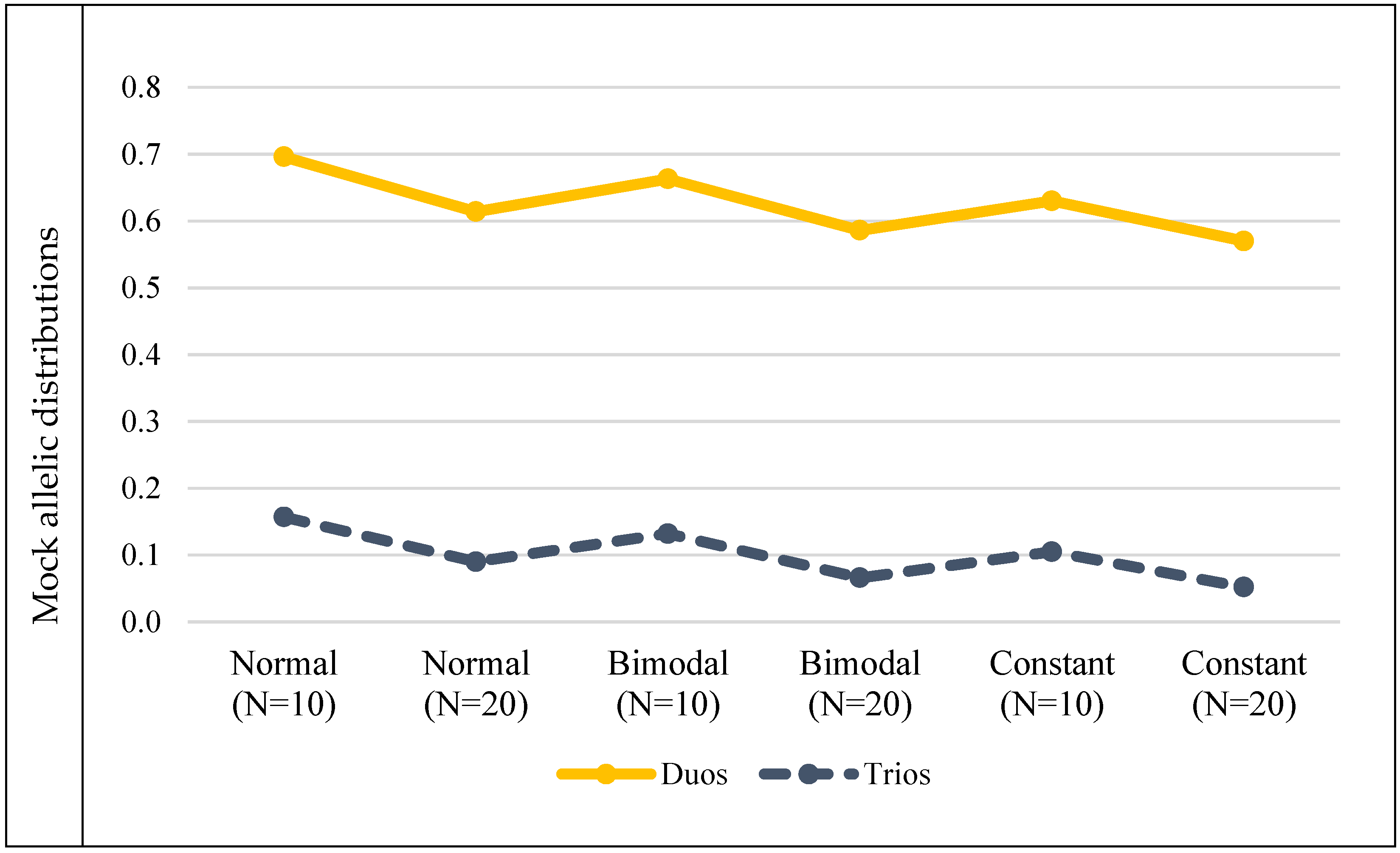
3.2. Mock Allele Frequency Distributions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Flores-Espinoza, R.; Paz-Cruz, E.; Ruiz-Pozo, V.A.; Lopez-Carrera, M.; Cabrera-Andrade, A.; Gusmão, L.; Burgos, G. Investigating genetic diversity in admixed populations from Ecuador. Am. J. Phys. Anthropol. 2021, 176, 109–119. [Google Scholar] [CrossRef]
- Srithawong, S.; Muisuk, K.; Srikummool, M.; Kampuansai, J.; Pittayaporn, P.; Ruangchai, S.; Liu, D.; Kutanan, W. Close genetic relationship between central Thai and Mon people in Thailand revealed by autosomal microsatellites. Int. J. Leg. Med. 2021, 135, 445–448. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Wang, Z.; Wang, M.; Hou, Y. Genetic Diversity and Phylogenetic Differentiation of Southwestern Chinese Han: A comprehensive and comparative analysis on 21 non-CODIS STRs. Sci. Rep. 2017, 7, 13730. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.X.; Mullikin, J.C.; Patterson, N.; Reich, D.E. Microsatellites are molecular clocks that support accurate inferences about history. Mol. Biol. Evol. 2009, 26, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, A.; Santos, C.; Pinheiro, M.; Pinto, P.; Soares, M.J.; Rocha, P.; Gusmão, L.; Amorim, A.; Van Der Hout, A.; Gerdes, A.-M.; et al. International distribution and age estimation of the Portuguese BRCA2 c.156_157insAlu founder mutation. Breast Cancer Res. Treat. 2011, 127, 671–679. [Google Scholar] [CrossRef]
- Sharony, R.; Martins, S.; Costa, I.P.D.; Zaltzman, R.; Amorim, A.; Sequeiros, J.; Gordon, C.R. Yemenite-Jewish families with Machado–Joseph disease (MJD/SCA3) share a recent common ancestor. Eur. J. Hum. Genet. 2019, 27, 1731. [Google Scholar] [CrossRef]
- Martins, S.; Soong, B.-W.; Wong, V.C.N.; Giunti, P.; Stevanin, G.; Ranum, L.P.W.; Sasaki, H.; Riess, O.; Tsuji, S.; Coutinho, P.; et al. Mutational origin of Machado-Joseph disease in the Australian Aboriginal communities of Groote Eylandt and Yirrkala. Arch. Neurol. 2012, 69, 746–751. [Google Scholar] [CrossRef]
- Martins, S.; Calafell, F.; Gaspar, C.; Wong, V.C.N.; Silveira, I.; Nicholson, G.A.; Brunt, E.R.; Tranebjaerg, L.; Stevanin, G.; Hsieh, M.; et al. Asian origin for the worldwide-spread mutational event in Machado-Joseph disease. Arch. Neurol. 2007, 64, 1502–1509. [Google Scholar] [CrossRef]
- Boattini, A.; Sarno, S.; Mazzarisi, A.M.; Viroli, C.; De Fanti, S.; Bini, C.; Larmuseau, M.H.D.; Pelotti, S.; Luiselli, D. Estimating Y-Str Mutation Rates and Tmrca Through Deep-Rooting Italian Pedigrees. Sci. Rep. 2019, 9, 9032. [Google Scholar] [CrossRef]
- Regueiro, M.; Alvarez, J.; Rowold, D.; Herrera, R.J. On the origins, rapid expansion and genetic diversity of Native Americans from hunting-gatherers to agriculturalists. Am. J. Phys. Anthr. 2013, 150, 333–348. [Google Scholar] [CrossRef]
- Strand, M.; Prolla, T.A.; Liskay, R.M.; Petes, T.D. Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature 1993, 365, 274–276. [Google Scholar] [CrossRef] [PubMed]
- Schlötterer, C.; Tautz, D. Slippage synthesis of simple sequence DNA. Nucleic Acids Res. 1992, 20, 211. [Google Scholar] [CrossRef] [PubMed]
- Eckert, K.A.; Hile, S.E. Every microsatellite is different: Intrinsic DNA features dictate mutagenesis of common microsatellites present in the human genome. Mol. Carcinog. 2009, 48, 379–388. [Google Scholar] [CrossRef]
- Wierdl, M.; Dominska, M.; Petes, T.D. Microsatellite instability in yeast: Dependence on the length of the microsatellite. Genetics 1997, 146, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, B.; Klintschar, M.; Neuhuber, F.; Hühne, J.; Rolf, B. Mutation Rate in Human Microsatellites: Influence of the Structure and Length of the Tandem Repeat. Am. J. Hum. Genet. 1998, 62, 1408–1415. [Google Scholar] [CrossRef]
- Ballantyne, K.N.; Goedbloed, M.; Fang, R.; Schaap, O.; Lao, O.; Wollstein, A.; Choi, Y.; van Duijn, K.; Vermeulen, M.; Brauer, S.; et al. Mutability of Y-Chromosomal Microsatellites: Rates, Characteristics, Molecular Bases, and Forensic Implications. Am. J. Hum. Genet. 2010, 87, 341. [Google Scholar] [CrossRef]
- Sun, J.X.; Helgason, A.; Masson, G.; Ebenesersdóttir, S.S.; Li, H.; Mallick, S.; Gnerre, S.; Patterson, N.; Kong, A.; Reich, D.; et al. A direct characterization of human mutation based on microsatellites. Nat. Genet. 2012, 44, 1161–1165. [Google Scholar] [CrossRef]
- Xu, X.; Peng, M.; Fang, Z.; Xu, X. The direction of microsatellite mutations is dependent upon allele length. Nat. Genet. 2000, 24, 396–399. [Google Scholar] [CrossRef]
- Weber, J.L.; Wong, C. Mutation of human short tandem repeats. Hum. Mol. Genet. 1993, 2, 1123–1128. [Google Scholar] [CrossRef]
- Kimura, M.; Ohta, T. Stepwise mutation model and distribution of allelic frequencies in a finite population. Proc. Natl. Acad. Sci. USA 1978, 75, 2868. [Google Scholar] [CrossRef]
- Pinto, N.; Gusmão, L.; Amorim, A. Mutation and mutation rates at y chromosome specific Short Tandem Repeat Polymorphisms (STRs): A reappraisal. Forensic Sci. Int. Genet. 2014, 9, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Antão-Sousa, S.; Sánchez-Diz, P.; Abovich, M.; Alvarez, J.; Carvalho, E.; Silva, C.; Domingues, P.; Farfán, M.; Gutierrez, A.; Pontes, L.; et al. Mutation rates and segregation data on 16 Y-STRs: An update to previous GHEP-ISFG studies. Forensic Sci. Int. Genet. Suppl. Ser. 2017, 6, e601–e602. [Google Scholar] [CrossRef][Green Version]
- Dupuy, B.M.; Stenersen, M.; Egeland, T.; Olaisen, B. Y-chromosomal microsatellite mutation rates: Differences in mutation rate between and within loci. Hum. Mutat. 2004, 23, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Su, Q.; Luo, H.; Li, Y.; Wu, J.; Yan, J.; Hou, Y.; Liang, W.; Zhang, L. Mutational analysis of 33 autosomal short tandem repeat (STR) loci in southwest Chinese Han population based on trio parentage testing. Forensic Sci. Int. Genet. 2016, 23, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Pinto, N.; Pereira, V.; Mas, C.T.; Loiola, S.; Carvalho, E.F.; Modesti, N.; Maxzud, M.; Marcucci, V.; Cano, H.; Cicarelli, R.; et al. Paternal and maternal mutations in X-STRs: A GHEP-ISFG collaborative study. Forensic Sci. Int. Genet. 2020, 46, 102258. [Google Scholar] [CrossRef]
- García, M.G.; Catanesi, C.I.; Penacino, G.A.; Gusmão, L.; Pinto, N. X-chromosome data for 12 STRs: Towards an Argentinian database of forensic haplotype frequencies. Forensic Sci. Int. Genet. 2019, 41, e8–e13. [Google Scholar] [CrossRef]
- Sun, H.; Liu, S.; Zhang, Y.; Whittle, M.R. Comparison of southern Chinese Han and Brazilian Caucasian mutation rates at autosomal short tandem repeat loci used in human forensic genetics. Int. J. Leg. Med. 2014, 128, 1–9. [Google Scholar] [CrossRef]
- Antão-Sousa, S.; Conde-Sousa, E.; Gusmão, L.; Amorim, A.; Pinto, N. Underestimation and misclassification of mutations at X chromosome STRs depend on population’s allelic profile. Forensic Science International: Genet. Suppl. Ser. 2019, 7, 718–720. [Google Scholar] [CrossRef]
- Slooten, K.; Ricciardi, F. Estimation of mutation probabilities for autosomal STR markers. Forensic Sci. Int. Genet. 2013, 7, 337–344. [Google Scholar] [CrossRef]
- Chakraborty, R.; Stivers, D.N.; Zhong, Y. Estimation of mutation rates from parentage exclusion data: Applications to STR and VNTR loci. Mutat. Res. 1996, 354, 41–48. [Google Scholar] [CrossRef]
- Vicard, P.; Dawid, A.P. A statistical treatment of biases affecting the estimation of mutation rates. Mutat. Res. 2004, 547, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.H. Multiple mutations, covert mutations and false exclusions in paternity casework. Int. Congr. Ser. 2004, 1261, 112–114. [Google Scholar] [CrossRef]
- Vicard, P.; Dawid, A.P.; Mortera, J.; Lauritzen, S.L. Estimating mutation rates from paternity casework. Forensic Sci. Int. Genet. 2008, 2, 9–18. [Google Scholar] [CrossRef]
- Kling, D.; Tillmar, A.O.; Egeland, T. Familias 3—Extensions and new functionality. Forensic Sci. Int. Genet. 2014, 13, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Lan, Q.; Wang, H.; Shen, C.; Guo, Y.; Yin, C.; Xie, T.; Fang, Y.; Zhou, Y.; Zhu, B. Mutability analysis towards 21 STR loci included in the AGCU 21 + 1 kit in Chinese Han population. Int. J. Leg. Med. 2018, 132, 1287–1291. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.L.; Chen, Y.F.; Huang, X.L.; Liu, K.Y.; Zhao, H.; Lu, D.J. Population data and mutation rates of 19 STR loci in seven provinces from China based on GoldeneyeTM DNA ID System 20A. Int. J. Leg. Med. 2017, 131, 653–656. [Google Scholar] [CrossRef] [PubMed]
- Hongdan, W.; Bing, K.; Ning, S.; Miao, H.; Bo, Z.; Yuxin, G.; Bofeng, Z.; Shixiu, L.; Zhaoshu, Z. Evaluation of the genetic parameters and mutation analysis of 22 STR loci in the central Chinese Han population. Int. J. Leg. Med. 2017, 131, 103–105. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, J.; Wang, H.; Liu, Z.-P.; Liu, M.; Zhang, Y.; Sun, L.; Zhang, H. Mutation rate estimation for 15 autosomal STR loci in a large population from Mainland China. Meta Gene 2015, 5, 150–156. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Markers | Duos | Trios | ||||||
|---|---|---|---|---|---|---|---|---|
| Norway | Somalia | Spain | Norway | Somalia | Spain | |||
| Allele Frequencies | Real allelic distributions | CSF1PO | 0.546 | 0.497 | 0.497 | 0.269 | 0.219 | 0.207 |
| D1S1656 | 0.249 | 0.312 | 0.325 | 0.087 | 0.128 | 0.135 | ||
| D21S11 | 0.372 | 0.348 | 0.35 | 0.163 | 0.15 | 0.147 | ||
| D2S441 | 0.497 | 0.419 | 0.393 | 0.141 | 0.125 | 0.108 | ||
| D3S1358 | 0.463 | 0.526 | 0.542 | 0.224 | 0.259 | 0.271 | ||
| FGA | 0.334 | 0.329 | 0.328 | 0.15 | 0.145 | 0.146 | ||
| SE33 | 0.143 | 0.178 | 0.171 | 0.054 | 0.069 | 0.065 | ||
| TH01 | 0.452 | 0.508 | 0.521 | 0.102 | 0.227 | 0.233 | ||
| TPOX | 0.622 | 0.508 | 0.525 | 0.119 | 0.192 | 0.198 | ||
| VWA | 0.445 | 0.425 | 0.435 | 0.213 | 0.205 | 0.205 | ||
| Mock allelic distributions | Normal (N = 10) | 0.696 | 0.157 | |||||
| Normal (N = 20) | 0.614 | 0.09 | ||||||
| Bimodal (N = 10) | 0.663 | 0.132 | ||||||
| Bimodal (N = 20) | 0.586 | 0.066 | ||||||
| Constant (N = 10) | 0.63 | 0.105 | ||||||
| Constant (N = 20) | 0.57 | 0.052 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antão-Sousa, S.; Conde-Sousa, E.; Gusmão, L.; Amorim, A.; Pinto, N. Estimations of Mutation Rates Depend on Population Allele Frequency Distribution: The Case of Autosomal Microsatellites. Genes 2022, 13, 1248. https://doi.org/10.3390/genes13071248
Antão-Sousa S, Conde-Sousa E, Gusmão L, Amorim A, Pinto N. Estimations of Mutation Rates Depend on Population Allele Frequency Distribution: The Case of Autosomal Microsatellites. Genes. 2022; 13(7):1248. https://doi.org/10.3390/genes13071248
Chicago/Turabian StyleAntão-Sousa, Sofia, Eduardo Conde-Sousa, Leonor Gusmão, António Amorim, and Nádia Pinto. 2022. "Estimations of Mutation Rates Depend on Population Allele Frequency Distribution: The Case of Autosomal Microsatellites" Genes 13, no. 7: 1248. https://doi.org/10.3390/genes13071248
APA StyleAntão-Sousa, S., Conde-Sousa, E., Gusmão, L., Amorim, A., & Pinto, N. (2022). Estimations of Mutation Rates Depend on Population Allele Frequency Distribution: The Case of Autosomal Microsatellites. Genes, 13(7), 1248. https://doi.org/10.3390/genes13071248