
Identification and Characterization of Variants in Intron 6 of the LPL Gene Locus among a Sample of the Kuwaiti Population


Abstract
1. Introduction
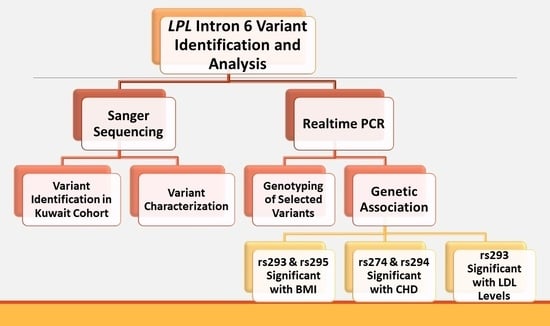
2. Materials and Methods
2.1. Study Population
2.2. Epidemiological Analysis
2.3. Sequencing Two Target Regions of Intron 6 at the LPL Gene Locus
2.4. Statistical Analysis
2.4.1. Determining the Genotype and Allele Frequencies
2.4.2. Testing the Association of the Detected InDels and SNPs with Phenotypic Variables
3. Results
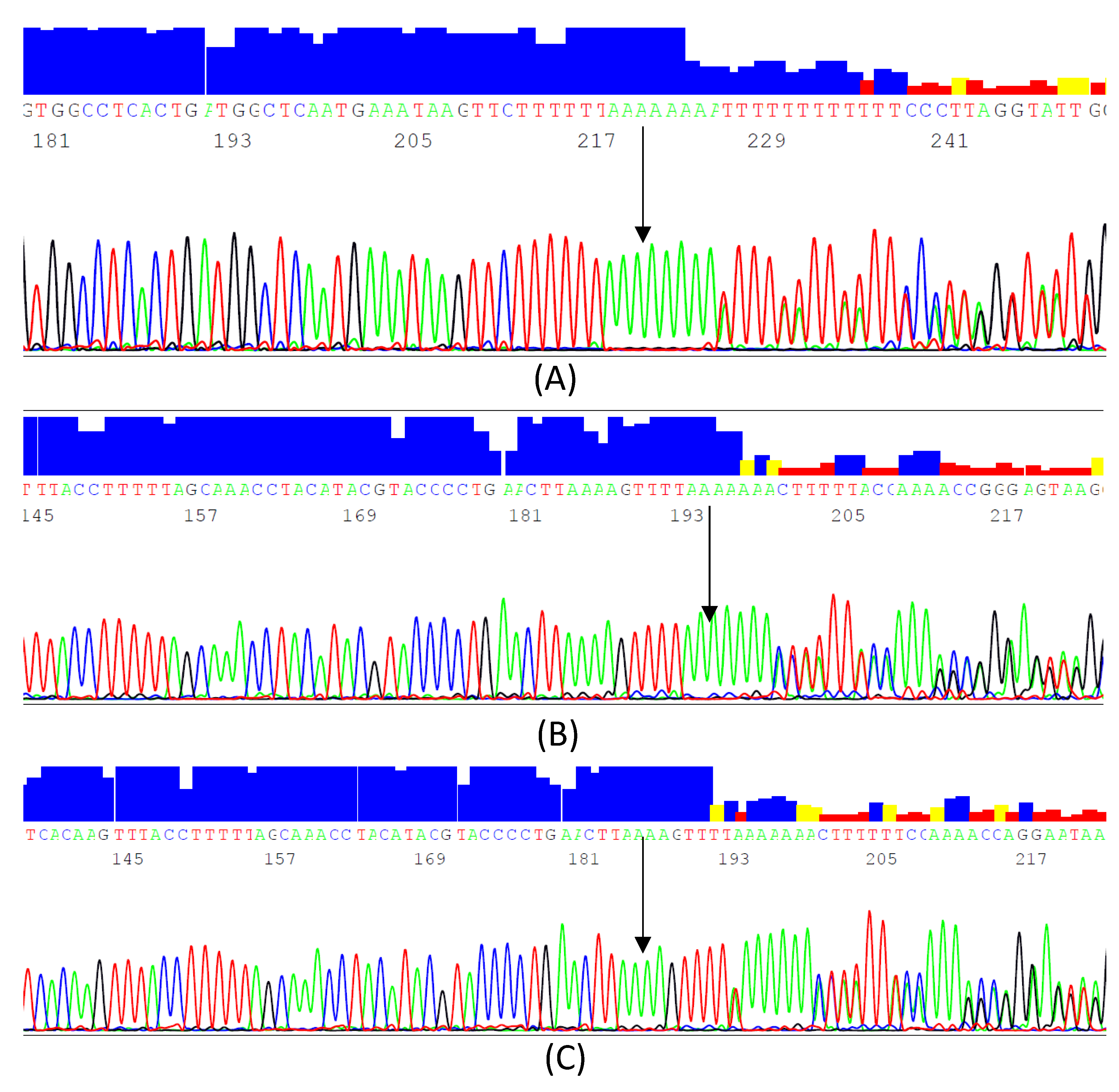
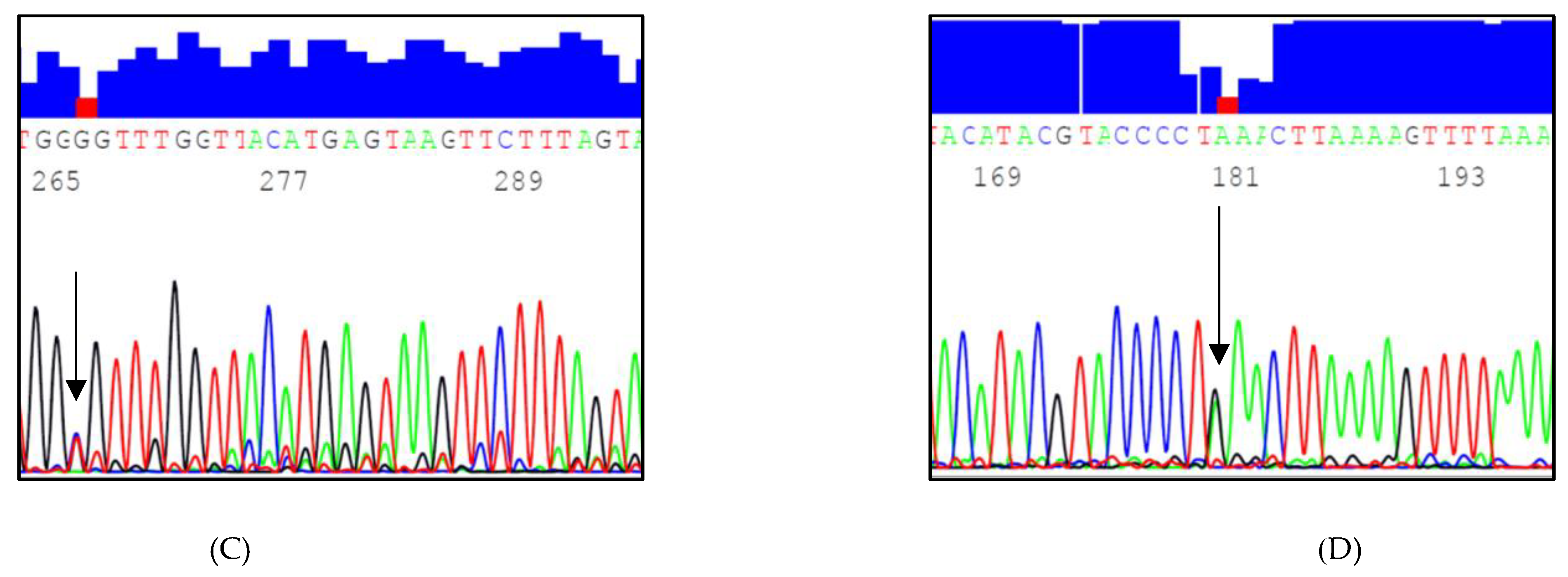
3.1. Identified InDels and SNPs
3.2. Genotype and Allele Frequencies of the Selected LPL Variants
3.3. Genetic Association with Lipid Levels
3.4. Genetic Association with BMI
3.5. Genetic Association with CHD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 5th ed.; W. H. Freeman: New York, NY, USA, 2002. Available online: https://www.ncbi.nlm.nih.gov/books/NBK21154/ (accessed on 1 October 2017).
- Kirchgessner, T.G.; LeBoeuf, R.C.; Langner, C.A.; Zollman, S.; Chang, C.H.; Taylor, B.A.; Schotz, M.C.; Gordon, J.I.; Lusis, A.J. Genetic and Developmental Regulation of the Lipoprotein Lipase Gene: Loci Both Distal and Proximal to the Lipoprotein Lipase Structural Gene Control Enzyme Expression. J. Biol. Chem. 1989, 264, 1473–1482. [Google Scholar] [CrossRef]
- Goldberg, I.J.; Eckel, R.H.; Abumrad, N.A. Regulation of Fatty Acid Uptake into Tissues: Lipoprotein Lipase- and CD36-Mediated Pathways. J. Lipid Res. 2009, 50, S86–S90. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.F.; Rader, D.J. New Insights into the Regulation of HDL Metabolism and Reverse Cholesterol Transport. Circ. Res. 2005, 96, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Apostol, G.; Schreiner, P.J.; Jacobs, D.R., Jr.; Boerwinkle, E.; Fornage, M. Associations of Lipoprotein Lipase Gene Polymorphisms with Longitudinal Plasma Lipid Trends in Young Adults: The Coronary Artery Risk Development in Young Adults (CARDIA) Study. Circ. Cardiovasc. Genet. 2010, 3, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Merkel, M.; Eckel, R.H.; Goldberg, I.J. Lipoprotein Lipase: Genetics, Lipid Uptake, and Regulation. J. Lipid Res. 2002, 43, 1997–2006. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Kreuzer, J.; Hamann, A.; Nawroth, P.P.; Dugi, K.A. The Proline 12 Alanine Substitution in the Peroxisome Proliferator--Activated Receptor-gamma2 Gene Is Associated with Lower Lipoprotein Lipase Activity In Vivo. Diabetes 2002, 51, 867–870. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.R.; Hooper, A.J.; Hegele, R.A. Familial Lipoprotein Lipase Deficiency. In Gene Reviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; University of Washington: Seattle, WA, USA, 2017; pp. 1993–2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1308/ (accessed on 1 May 2018).
- Klos, K.L.; Kullo, I.J. Genetic Determinants of HDL: Monogenic Disorders and Contributions to Variation. Curr. Opin. Cardiol. 2007, 22, 344–351. [Google Scholar] [CrossRef]
- Xie, L.; Li, Y.M. Lipoprotein Lipase (LPL) Polymorphism and the Risk of Coronary Artery Disease: A Meta-Analysis. Int. J. Environ. Res. Public Health 2017, 14, 84. [Google Scholar] [CrossRef] [PubMed]
- Bogari, N.M.; Aljohani, A.; Dannoun, A.; Elkhateeb, O.; Porqueddu, M.; Amin, A.A.; Bogari, D.N.; Taher, M.M.; Buba, F.; Allam, R.M.; et al. Association Between HindIII (rs320) Variant in the Lipoprotein Lipase Gene and the Presence of Coronary Artery Disease and Stroke Among the Saudi Population. Saudi J. Biol. Sci. 2020, 27, 2018–2024. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Tortosa, D.F.; Pascual-Gamarra, J.M.; Labayen, I.; Rupérez, A.I.; Censi, L.; Béghin, L.; Michels, N.; Gonzalez-Gross, M.; Manios, Y.; Lambrinou, C.P.; et al. Association Between Lipoprotein Lipase Gene Polymorphisms and Cardiovascular Disease Risk Factors in European Adolescents: The Healthy Lifestyle in Europe by Nutrition in Adolescence Study. Pediatr. Diabetes 2020, 21, 747–757. [Google Scholar] [CrossRef]
- Deeb, S.S.; Peng, R.L. Structure of the Human Lipoprotein Lipase Gene. Biochemistry 1989, 28, 4131–4135. [Google Scholar] [CrossRef] [PubMed]
- Sparkes, R.S.; Zollman, S.; Klisak, I.; Kirchgessner, T.G.; Komaromy, M.C.; Mohandas, T.; Schotz, M.C.; Lusis, A.J. Human Genes Involved in Lipolysis of Plasma Lipoproteins: Mapping of Loci for Lipoprotein Lipase to 8p22 and Hepatic Lipase to 15q21. Genomics 1987, 1, 138–144. [Google Scholar] [CrossRef]
- Lo, J.Y.; Smith, L.C.; Chan, L. Lipoprotein Lipase: Role of Intramolecular Disulfide Bonds in Enzyme Catalysis. Biochem. Biophys. Res. Commun. 1995, 206, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Van Tilbeurgh, H.; Roussel, A.; Lalouel, J.M.; Cambillau, C. Lipoprotein Lipase. Molecular Model Based on the Pancreatic Lipase X-Ray Structure: Consequences for Heparin Binding and Catalysis. J. Biol. Chem. 1994, 269, 4626–4633. [Google Scholar] [CrossRef]
- Li, Y.; He, P.P.; Zhang, D.W.; Zheng, X.L.; Cayabyab, F.S.; Yin, W.D.; Tang, C.K. Lipoprotein Lipase: From Gene to Atherosclerosis. Atherosclerosis 2014, 237, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Stein, Y.; Stein, O. Lipoprotein Lipase and Atherosclerosis. Atherosclerosis 2003, 170, 1–9. [Google Scholar] [CrossRef]
- Glock, B.; Schwartz, D.W.; Schwartz-Jungl, E.M.; Mayr, W.R. Allelic Ladder Characterization of the Short Tandem Repeat Polymorphism in intron 6 of the Lipoprotein Lipase Gene and Its Application in an Austrian Caucasian Population Study. J. Forensic Sci. 1996, 41, 579–581. [Google Scholar] [CrossRef]
- Maruyama, S.; Minaguchi, K. Polymorphism of LPL Locus in Japanese and Comparison of PCR Amplification Efficiency from Degraded DNA Between LPL Locus and the D21S11. Bull. Tokyo Dent. Coll. 2005, 46, 115–121. [Google Scholar] [CrossRef][Green Version]
- Goodarzi, M.O.; Wong, H.; Quiñones, M.J.; Taylor, K.D.; Guo, X.; Castellani, L.W.; Antoine, H.J.; Yang, H.; Hsueh, W.A.; Rotter, J.I. The 3′ Untranslated Region of the Lipoprotein Lipase Gene: Haplotype Structure and Association with Post-Heparin Plasma Lipase Activity. J. Clin. Endocrinol. Metab. 2005, 90, 4816–4823. [Google Scholar] [CrossRef]
- Pirim, D.; Wang, X.; Radwan, Z.H.; Niemsiri, V.; Hokanson, J.E.; Hamman, R.F.; Barmada, M.M.; Demirci, F.Y.; Kamboh, M.I. Lipoprotein Lipase Gene Sequencing and Plasma Lipid Profile. J. Lipid Res. 2014, 55, 85–93. [Google Scholar] [CrossRef]
- Al-Bustan, S.A.; Al-Serri, A.; Annice, B.G.; Alnaqeeb, M.A.; Al-Kandari, W.Y.; Dashti, M. A Novel LPL Intronic Variant: G.18704C>A Identified by Re-Sequencing Kuwaiti Arab Samples Is Associated with High-Density Lipoprotein, Very Low-Density Lipoprotein and Triglyceride Lipid Levels. PLoS ONE 2018, 13, e0192617. [Google Scholar] [CrossRef] [PubMed]
- Kraja, A.T.; Vaidya, D.; Pankow, J.S.; Goodarzi, M.O.; Assimes, T.L.; Kullo, I.J.; Sovio, U.; Mathias, R.A.; Sun, Y.V.; Franceschini, N.; et al. A Bivariate Genome-Wide Approach to Metabolic Syndrome: STAMPEED Consortium. Diabetes 2011, 60, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Pirim, D.; Wang, X.; Radwan, Z.H.; Niemsiri, V.; Bunker, C.H.; Barmada, M.M.; Kamboh, M.I.; Demirci, F.Y. Resequencing of LPL in African Blacks and Associations with Lipoprotein-Lipid Levels. Eur. J. Hum. Genet. 2015, 23, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.D.; Scheuner, M.T.; Yang, H.; Wang, Y.; Haritunians, T.; Fischel-Ghodsian, N.; Shah, P.K.; Forrester, J.S.; Knatterud, G.; Rotter, J.I. Lipoprotein Lipase Locus and Progression of Atherosclerosis in Coronary-Artery Bypass Grafts. Genet. Med. 2004, 6, 481–486. [Google Scholar] [CrossRef][Green Version]
- Shimada, M.; Ishibashi, S.; Inaba, T.; Yagyu, H.; Harada, K.; Osuga, J.I.; Ohashi, K.; Yazaki, Y.; Yamada, N. Suppression of Diet-Induced Atherosclerosis in Low Density Lipoprotein Receptor Knockout Mice Overexpressing Lipoprotein Lipase. Proc. Natl. Acad. Sci. USA 1996, 93, 7242–7246. [Google Scholar] [CrossRef]
- Zilversmit, D.B. A Proposal Linking Atherogenesis to the Interaction of Endothelial Lipoprotein Lipase with Triglyceride-Rich Lipoproteins. Circ. Res. 1973, 33, 633–638. [Google Scholar] [CrossRef]
- Ichikawa, T.; Kitajima, S.; Liang, J.; Koike, T.; Wang, X.; Sun, H.; Okazaki, M.; Morimoto, M.; Shikama, H.; Watanabe, T.; et al. Overexpression of Lipoprotein Lipase in Transgenic Rabbits Leads to Increased Small Dense LDL in the Plasma and Promotes Atherosclerosis. Lab. Investig. 2004, 84, 715–726. [Google Scholar] [CrossRef][Green Version]
- Auwerx, J.H.; Deeb, S.; Brunzell, J.D.; Peng, R.; Chait, A. Transcriptional Activation of the Lipoprotein Lipase and Apolipoprotein E Genes Accompanies Differentiation in Some Human Macrophage-Like Cell Lines. Biochemistry 1988, 27, 2651–2655. [Google Scholar] [CrossRef]
- Glass, C.K.; Witztum, J.L. Atherosclerosis. The Road Ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef]
- Kumari, A.; Kristensen, K.K.; Ploug, M.; Winther, A.L. The Importance of Lipoprotein Lipase Regulation in Atherosclerosis. Biomedicines 2021, 9, 782. [Google Scholar] [CrossRef]
- Feingold, K.R. Introduction to Lipids and Lipoproteins. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2015; p. 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK305896/ (accessed on 1 May 2018).
- Klop, B.; Elte, J.W.; Cabezas, M.C. Dyslipidemia in Obesity: Mechanisms and Potential Targets. Nutrients 2013, 5, 1218–1240. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, E.A.; Myasoedova, V.A.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. Small Dense Low-Density Lipoprotein as Biomarker for Atherosclerotic Diseases. Oxid. Med. Cell. Longev. 2017, 2017, 1273042. [Google Scholar] [CrossRef] [PubMed]
- Hokanson, J.E.; Brunzell, J.D.; Jarvik, G.P.; Wijsman, E.M.; Austin, M.A. Linkage of Low-Density Lipoprotein Size to the Lipoprotein Lipase Gene in Heterozygous Lipoprotein Lipase Deficiency. Am. J. Hum. Genet. 1999, 64, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Ruel, I.L.; Gaudet, D.; Perron, P.; Bergeron, J.; Julien, P.; Lamarche, B. Characterization of LDL Particle Size Among Carriers of a Defective or a Null Mutation in the Lipoprotein Lipase Gene: The Québec LIPD Study. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1181–1186. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Garcia-Arcos, I.; Hiyama, Y.; Drosatos, K.; Bharadwaj, K.G.; Hu, Y.; Son, N.H.; O’Byrne, S.M.; Chang, C.L.; Deckelbaum, R.J.; Takahashi, M.; et al. Adipose-Specific Lipoprotein Lipase Deficiency More Profoundly Affects Brown Fat Biology than White Fat. J. Biol. Chem. 2013, 288, 14046–14058. [Google Scholar] [CrossRef]
- Jensen, D.R.; Schlaepfer, I.R.; Morin, C.L.; Pennington, D.S.; Marcell, T.; Ammon, S.M.; Gutierrez-Hartmann, A.; Eckel, R.H. Prevention of Diet-Induced Obesity in Transgenic Mice Overexpressing Skeletal Muscle Lipoprotein Lipase. Am. J. Physiol. 1997, 273, R683–R689. [Google Scholar] [CrossRef]
- Hara, T.; Cameron-Smith, D.; Cooney, G.J.; Kusunoki, M.; Tsutsumi, K.; Storlien, L.H. The Actions of a Novel Lipoprotein Lipase Activator, NO-1886, in Hypertriglyceridemic Fructose-Fed Rats. Metabolism 1998, 47, 149–153. [Google Scholar] [CrossRef]
- Kern, P.A.; Ong, J.M.; Saffari, B.; Carty, J. The Effect of Weight Loss on the Activity and Expression of Adipose Tissue Lipoprotein Lipase in Very Obese Humans. N. Engl. J. Med. 1990, 322, 1053–1059. [Google Scholar] [CrossRef]
- Ferland, A.; Château-Degat, M.L.; Hernandez, T.L.; Eckel, R.H. Tissue-Specific Responses of Lipoprotein Lipase to Dietary Macronutrient Composition as a Predictor of Weight Gain Over 4 Years. Obesity 2012, 20, 1006–1011. [Google Scholar] [CrossRef]
- Zheng, Q.; Ma, Y.; Chen, S.; Che, Q.; Chen, D. The Integrated Landscape of Biological Candidate Causal Genes in Coronary Artery Disease. Front. Genet. 2020, 11, 320. [Google Scholar] [CrossRef]
- Ahmadi, Z.; Senemar, S.; Toosi, S.; Radmanesh, S. The Association of Lipoprotein Lipase Genes, HindIII, and S447X Polymorphisms with Coronary Artery Disease in Shiraz City. J. Cardiovasc. Thorac. Res. 2015, 7, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Sagoo, G.S.; Tatt, I.; Salanti, G.; Butterworth, A.S.; Sarwar, N.; van Maarle, M.; Jukema, J.W.; Wiman, B.; Kastelein, J.J.; Bennet, A.M.; et al. Seven Lipoprotein Lipase Gene Polymorphisms, Lipid Fractions, and Coronary Disease: A HuGE Association Review and Meta-Analysis. Am. J. Epidemiol. 2008, 168, 1233–1246. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; King, G.J.; Bair, T.L.; Elmer, S.P.; Muhlestein, J.B.; Habashi, J.; Mixson, L.; Carlquist, J.F. Association of Lipoprotein Lipase Gene Polymorphisms with Coronary Artery Disease. J. Am. Coll. Cardiol. 1999, 33, 1013–1020. [Google Scholar] [CrossRef]
- Rip, J.; Nierman, M.C.; Wareham, N.J.; Luben, R.; Bingham, S.A.; Day, N.E.; van Miert, J.N.; Hutten, B.A.; Kastelein, J.J.; Kuivenhoven, J.A.; et al. Serum Lipoprotein Lipase Concentration and Risk for Future Coronary Artery Disease: The EPIC-Norfolk Prospective Population Study. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Won, H.H.; Peloso, G.M.; O’Dushlaine, C.; Liu, D.; Stitziel, N.O.; Natarajan, P.; Nomura, A.; Emdin, C.A.; Gupta, N.; et al. Association of Rare and Common Variation in the Lipoprotein Lipase Gene with Coronary Artery Disease. JAMA 2017, 317, 937–946. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subjects | |||
|---|---|---|---|
| Gender | Males | Females | Total |
| Number | 372 | 357 | 729 |
| Age (Mean ± Std) | 44.8 ± 16.1 | 44.2 ± 14.5 | 44.5 ± 15.3 |
| BMI n (%) | |||
| <25 | 80 (21.7) | 50 (14) | 130 (17.9) |
| 25–30 | 135 (36.6) | 96 (27) | 231 (31.9) |
| >30 | 154 (41.7) | 210 (59) | 364 (50.2) |
| Variant | Location | Type | MAF | Global MAF * | Predicted Consequences | Number of Subjects | HWE |
|---|---|---|---|---|---|---|---|
| rs293 | Chr8:19958568-19958575 c.1019-685_1019-684insA | Insertion A | 0.15 | 0.25 | Intronic variant | 177 | <0.001 |
| rs274 | Chr8:19956466-19956469 c.1018+386_1018+387insA | Insertion A | 0.06 | 0.07 | Intronic variant | 93 | 0.178 |
| rs294 | Chr8:19958614 c.1019-646T>C | SNP (T>C) | 0.07 | 0.13 | Intronic variant | 80 | <0.001 |
| rs295 | Chr8:19958727 c.1019-533A>C | SNP (A>C) | 0.12 | 0.27 | Intronic variant | 139 | <0.001 |
| rs144578061 | Chr8:19956367 c.1018+284A>C | SNP (A>C) | 0.03 | <0.01 | Intronic variant | 37 | 0.483 |
| rs74746426 | Chr8:19958433 c.1019-827A>T | SNP (A>T) | 0.02 | 0.02 | Intronic variant | 23 | 0.662 |
| rs296 | Chr8:19958733c.1019-527G>A | SNP (G>A) | <0.01 | <0.01 | Intronic variant | 2 | 1 |
| rs901601579 | Chr8:19958719c.1019-541G>A | SNP (G>A) | <0.01 | Not available | Intronic variant | 1 | 1 |
| rs138618627 | Chr8:19956378c.1018+295C>A | SNP (C>A) | <0.01 | <0.01 | Intronic variant | 1 | 1 |
| rs272 | Chr8:19956417c.1018+334C>G | SNP (C>G) | <0.01 | 0.02 | Intronic variant | 4 | 0.920 |
| rs540562340 | Chr8:19956532c.1018+449T>G | SNP (T>G) | <0.01 | <0.01 | Intronic variant | 1 | 1 |
| Novel rs294 genotype | Chr8:19958614 c.1019-646T>G | SNP (T>G) | 0.01 | Not available | Intronic variant | 10 | 0.862 |
| Novel SNP | Chr8:19956480 c.1018+397C>T | SNP (C>T) | <0.01 | Not available | Intronic variant | 1 | 1 |
| rs292 | Chr8:19958544 c.1019-716G>A | SNP (G>A) | <0.01 | <0.01 | Intronic variant | 1 | 1 |
| rs910411725 | Chr8:19958707 c.1019-553A>G | SNP (A>G) | <0.01 | <0.01 | Intronic variant | 1 | 1 |
| rs297 | Chr8:19958860c.1019-400T>C | SNP (T>C) | <0.01 | 0.25 | Intronic variant | 6 | 0.92 |
| Variable | Total n = 729 n (%) |
|---|---|
| rs274 | |
| No insertion | 636 (87.2) |
| Heterozygote insertion | 81 (11.1) |
| Homozygote insertion | 5 (0.7) |
| Allele frequencies wildtype/insertion | 0.94/0.06 |
| p-value HWE | 0.178 |
| rs293 | |
| AA (wildtype) | 552 (75.7) |
| Aa (Heterozygote mutant) | 139 (19.1) |
| aa (Homozygote mutant) | 38 (5.2) |
| Allele frequencies A/a | 0.85/0.15 |
| p-value HWE | <0.001 |
| rs294 | |
| TT (wildtype) | 649 (89) |
| TC (Heterozygote mutant) | 63 (8.6) |
| CC (Homozygote mutant) | 17 (2.3) |
| Allele frequencies T/C | 0.93/0.07 |
| p-value HWE | <0.001 |
| rs295 | |
| AA (wildtype) | 590 (80.9) |
| AC (Heterozygote mutant) | 99 (13.6) |
| CC (Homozygote mutant) | 40 (5.5) |
| Allele frequencies T/C | 0.88/0.12 |
| p-value HWE | <0.001 |
| SNP | Variable | W/W | n | W/M + M/M | n | p-Value |
|---|---|---|---|---|---|---|
| rs274 | TC | 4.90 ± 1.02 | 389 | 4.92 ± 0.88 | 77 | 0.904 |
| TG | 1.31 ± 0.98 | 389 | 1.24 ± 0.54 | 77 | 0.555 | |
| HDL | 1.06 ± 0.39 | 389 | 1.12 ± 0.42 | 77 | 0.213 | |
| VLDL | 0.55 ± 0.43 | 389 | 0.53 ± 0.24 | 77 | 0.626 | |
| LDL | 3.21 ± 0.90 | 389 | 3.22 ± 0.79 | 77 | 0.885 | |
| rs293 | TC | 4.85 ± 0.99 | 312 | 5.02 ± 1.01 | 154 | 0.092 |
| TG | 1.35 ± 1.01 | 312 | 1.19 ± 0.70 | 154 | 0.069 | |
| HDL | 1.05 ± 0.37 | 312 | 1.11 ± 0.42 | 154 | 0.133 | |
| VLDL | 0.57 ± 0.44 | 312 | 0.50 ± 0.30 | 154 | 0.058 | |
| LDL | 3.14 ± 0.88 | 312 | 3.34 ± 0.86 | 154 | 0.024 * | |
| rs294 | TC | 4.88 ± 0.96 | 406 | 5.10 ± 1.21 | 60 | 0.099 |
| TG | 1.30 ± 0.94 | 406 | 1.26 ± 0.80 | 60 | 0.736 | |
| HDL | 1.06 ± 0.39 | 406 | 1.15 ± 0.39 | 60 | 0.090 | |
| VLDL | 0.55 ± 0.41 | 406 | 0.53 ± 0.34 | 60 | 0.693 | |
| LDL | 3.18 ± 0.86 | 406 | 3.38 ± 1.03 | 60 | 0.111 | |
| rs295 | TC | 4.87 ± 0.98 | 350 | 5.02 ± 1.04 | 116 | 0.156 |
| TG | 1.32 ± 0.97 | 350 | 1.21 ± 0.75 | 116 | 0.257 | |
| HDL | 1.06 ± 0.39 | 350 | 1.11 ± 0.41 | 116 | 0.237 | |
| VLDL | 0.56 ± 0.43 | 350 | 0.51 ± 0.32 | 116 | 0.210 | |
| LDL | 3.16 ± 0.87 | 350 | 3.34 ± 0.89 | 116 | 0.059 |
| Lipid | Variable | β-Coefficient | Lower 95% CI | Upper 95% CI | p-Value * |
|---|---|---|---|---|---|
| TC | rs293 | 0.167 | −0.026 | 0.360 | 0.090 |
| Age | 0.001 | −0.005 | 0.007 | 0.697 | |
| Sex (Female) | 0.008 | −0.177 | 0.192 | 0.936 | |
| BMI | 0.004 | −0.007 | 0.016 | 0.464 | |
| TG | rs293 | −0.078 | −0.185 | 0.030 | 0.157 |
| Age | 0.008 | 0.005 | 0.012 | <0.0001 | |
| Sex (Female) | −0.182 | −0.284 | −0.079 | 0.001 | |
| BMI | 0.014 | 0.007 | 0.020 | <0.0001 | |
| HDL | rs293 | 0.044 | −0.041 | 0.128 | 0.309 |
| Age | −0.001 | −0.004 | 0.001 | 0.368 | |
| Sex (Female) | 0.232 | 0.152 | 0.313 | <0.0001 | |
| BMI | −0.004 | −0.009 | 0.001 | 0.096 | |
| VLDL | rs293 | −0.087 | −0.197 | 0.023 | 0.122 |
| Age | 0.008 | 0.005 | 0.012 | <0.0001 | |
| Sex (Female) | −0.171 | −0.277 | −0.066 | 0.001 | |
| BMI | 0.013 | 0.006 | 0.019 | 0.0001 | |
| LDL | rs293 | 0.191 | 0.021 | 0.360 | 0.027 |
| Age | −0.003 | −0.008 | 0.003 | 0.308 | |
| Sex (Female) | −0.145 | −0.307 | 0.017 | 0.080 |
| SNP | W/W | n | W/M | n | M/M | n | Kruskal-Wallis p-Value | β- Coefficient (Recessive Model) | 95% CI (Recessive Model) | p-Value (Recessive Model) |
|---|---|---|---|---|---|---|---|---|---|---|
| rs274 | 31.34 ± 7.63 | 633 | 33.18 ± 7.84 | 83 | 30.65 ± 7.98 | 9 | 0.091 | _ | _ | _ |
| rs293 | 31.31 ± 7.54 | 549 | 31.30 ± 7.23 | 138 | 35.80 ± 9.89 | 38 | 0.015 * | 4.032 | 1.572 –6.491 | 0.001 * |
| rs294 | 31.53 ± 7.67 | 645 | 30.39 ± 6.28 | 63 | 36.27 ± 10.85 | 17 | 0.102 | − | − | − |
| rs295 | 31.40 ± 7.55 | 587 | 30.89 ± 7.24 | 98 | 35.18 ± 9.52 | 40 | 0.024 * | 3.318 | 0.906 –5.729 | 0.007 * |
| Control (n = 508) | Case (n = 217) | OR (95% CI) | p-Value | |
|---|---|---|---|---|
| Codominant Model | ||||
| −/− | 65.5% (333) | 99.5% (216) | 18.125 (2.395–137.176) | 0.005 |
| −/A | 27.2% (138) | 0 | <0.0001 | 0.996 |
| A/A | 7.3% (37) | 0.5% (1) | 1 | |
| Dominant Model | ||||
| −/− | 65.5% (333) | 99.5% (216) | 1 | |
| −/A + A/A | 34.5% (175) | 0.5% (1) | 0.009 (0.001–0.068) | <0.0001 |
| Recessive Model | ||||
| −/− + −/A | 92.7% (471) | 99.5% (216) | 1 | |
| A/A | 7.3% (37) | 0.5% (1) | 0.078 (0.010–0.591) | 0.013 |
| Control (n = 508) | Case (n = 217) | OR (95% CI) | p-Value | |
|---|---|---|---|---|
| Codominant Model | ||||
| TT | 86.0% (437) | 95.9% (208) | 1 | |
| TC | 11.4% (58) | 2.3% (5) | 0.194 (0.072–0.526) | 0.001 |
| CC | 2.6% (13) | 1.8% (4) | 1.025 (0.290–3.624) | 0.970 |
| Dominant Model | ||||
| TT | 86.0% (437) | 95.9% (208) | 1 | |
| TC+CC | 14.0% (71) | 4.1% (9) | 0.316 (0.146–0.686) | 0.004 |
| Recessive Model | ||||
| TT+TC | 97.4% (495) | 98.2% (213) | 1 | |
| CC | 2.6% (13) | 1.8% (4) | 1.115 (0.315–3.953) | 0.866 |
| Control (n = 508) | Case (n = 217) | OR (95% CI) | p-Value | |
|---|---|---|---|---|
| Dominant Model | ||||
| −/− | 83.3% (423) | 96.8% (210) | 1 | |
| −/A,T + A,T/A,T | 16.7% (85) | 3.2% (7) | 0.179 (0.077–0.414) | <0.0001 |
| Variant | Kuwait | European (Non-Finnish) | European (Finnish) | African /African American | Latino/ Admixed American | East Asian | South Asian | Ashk-Enazi Jews |
|---|---|---|---|---|---|---|---|---|
| rs274 | 0.060 | 0.0004 | N/A | 0.0681 | N/A | 0.0000 | N/A | 0.0111 |
| rs293 | 0.150 | 0.2614 * | N/A | 0.3169 * | N/A | 0.2341 * | 0.208 * | N/A |
| rs294 | 0.070 | 0.1401 | 0.1313 | 0.1445 | 0.1580 | 0.1001 | 0.000 | 0.1840 |
| rs295 | 0.120 | 0.2421 | 0.2301 | 0.3745 | 0.2298 | 0.2033 | 0.000 | 0.3586 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Shammari, R.T.; Al-Serri, A.E.; Barhoush, S.A.; Al-Bustan, S.A. Identification and Characterization of Variants in Intron 6 of the LPL Gene Locus among a Sample of the Kuwaiti Population. Genes 2022, 13, 664. https://doi.org/10.3390/genes13040664
Al-Shammari RT, Al-Serri AE, Barhoush SA, Al-Bustan SA. Identification and Characterization of Variants in Intron 6 of the LPL Gene Locus among a Sample of the Kuwaiti Population. Genes. 2022; 13(4):664. https://doi.org/10.3390/genes13040664
Chicago/Turabian StyleAl-Shammari, Reem T., Ahmad E. Al-Serri, Sahar A. Barhoush, and Suzanne A. Al-Bustan. 2022. "Identification and Characterization of Variants in Intron 6 of the LPL Gene Locus among a Sample of the Kuwaiti Population" Genes 13, no. 4: 664. https://doi.org/10.3390/genes13040664
APA StyleAl-Shammari, R. T., Al-Serri, A. E., Barhoush, S. A., & Al-Bustan, S. A. (2022). Identification and Characterization of Variants in Intron 6 of the LPL Gene Locus among a Sample of the Kuwaiti Population. Genes, 13(4), 664. https://doi.org/10.3390/genes13040664