
Transcriptomic Adjustments in a Freshwater Ectoparasite Reveal the Role of Molecular Plasticity for Parasite Host Shift

 ,
,  ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. Study Model
2.2. Sampling Design and Sequencing
2.3. Gene Expression Analyses
2.3.1. Quantification of Gene Expression Levels
2.3.2. Identification of Differentially Expressed Genes
2.3.3. Functional Analyses
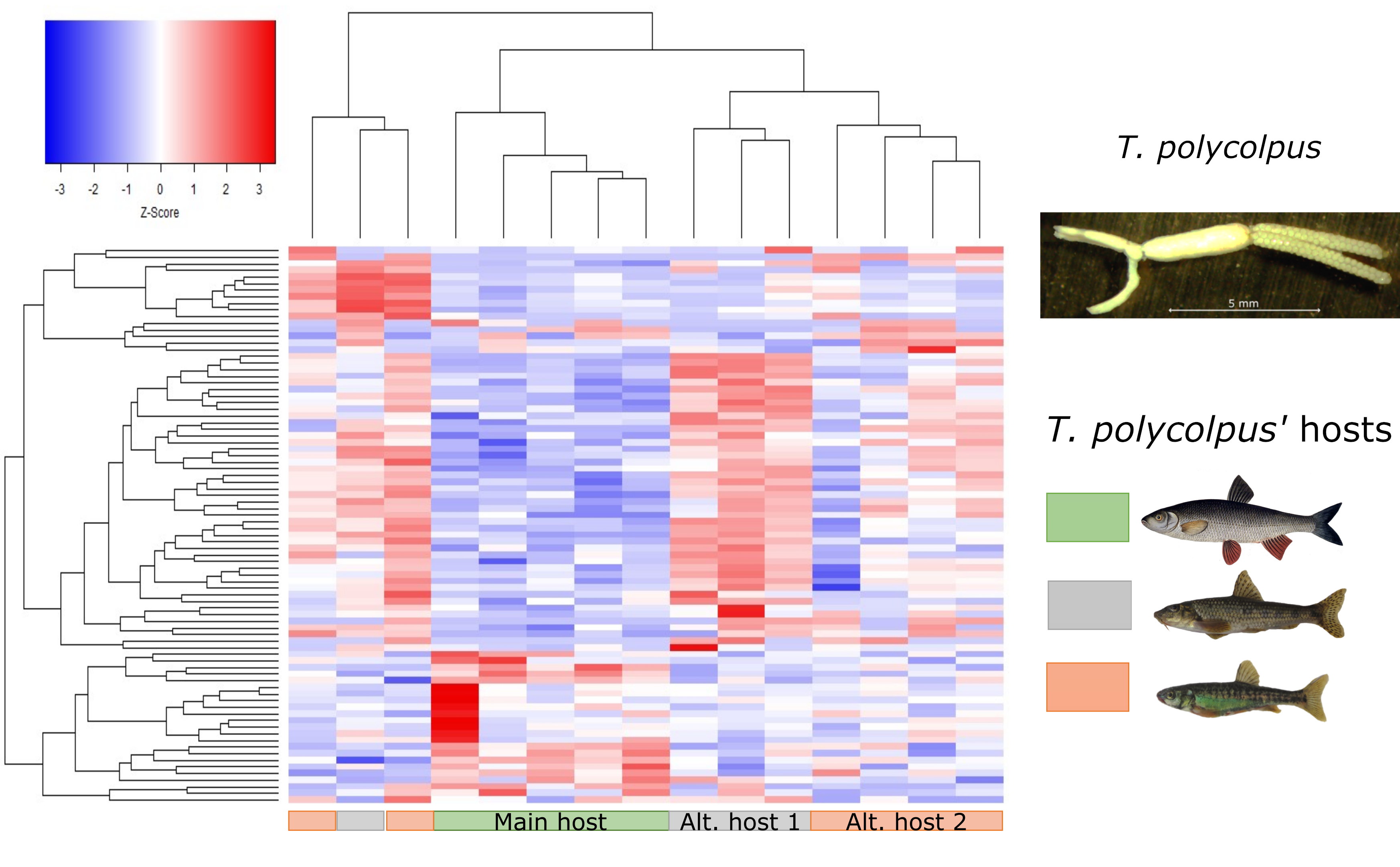
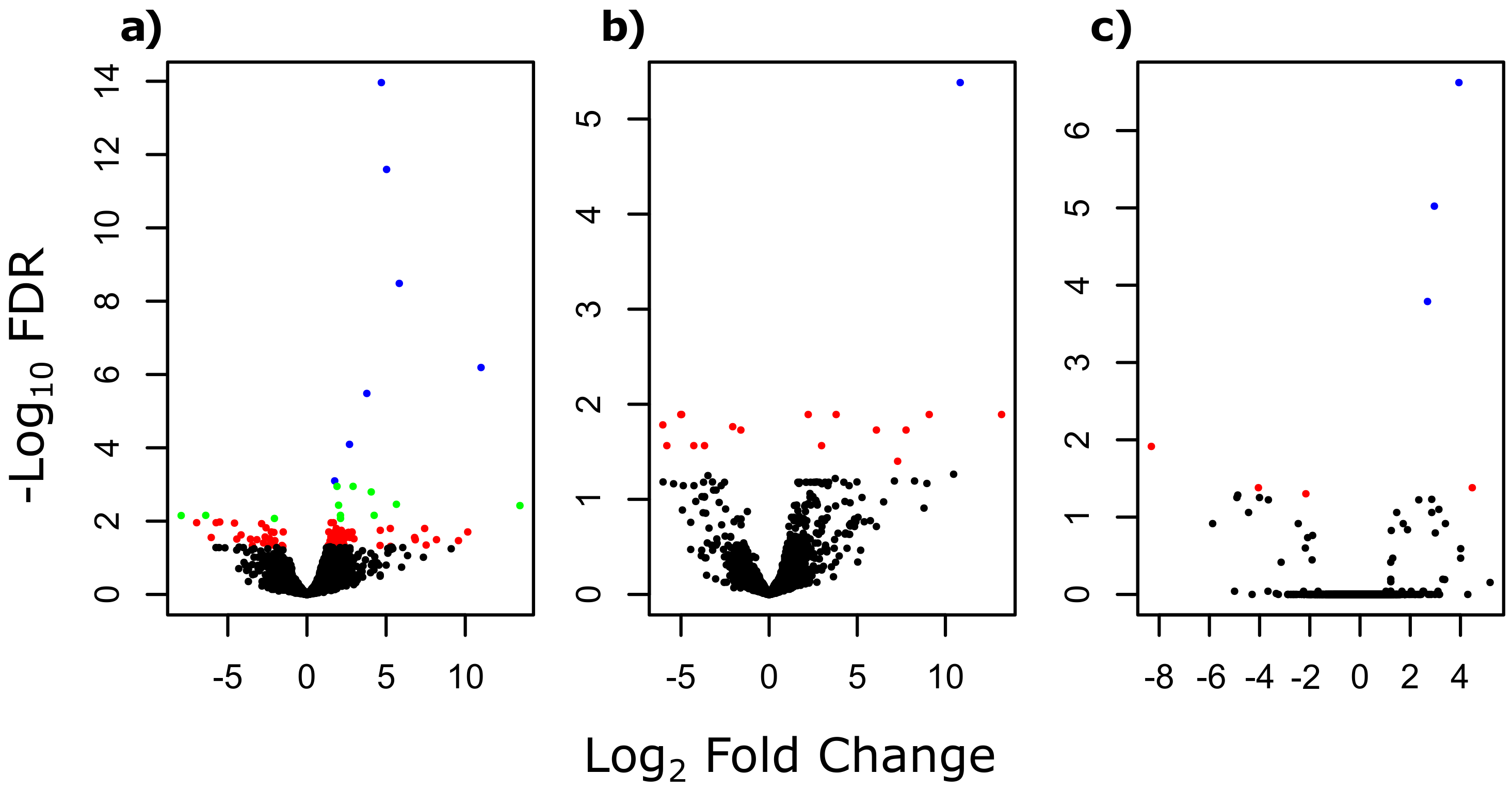
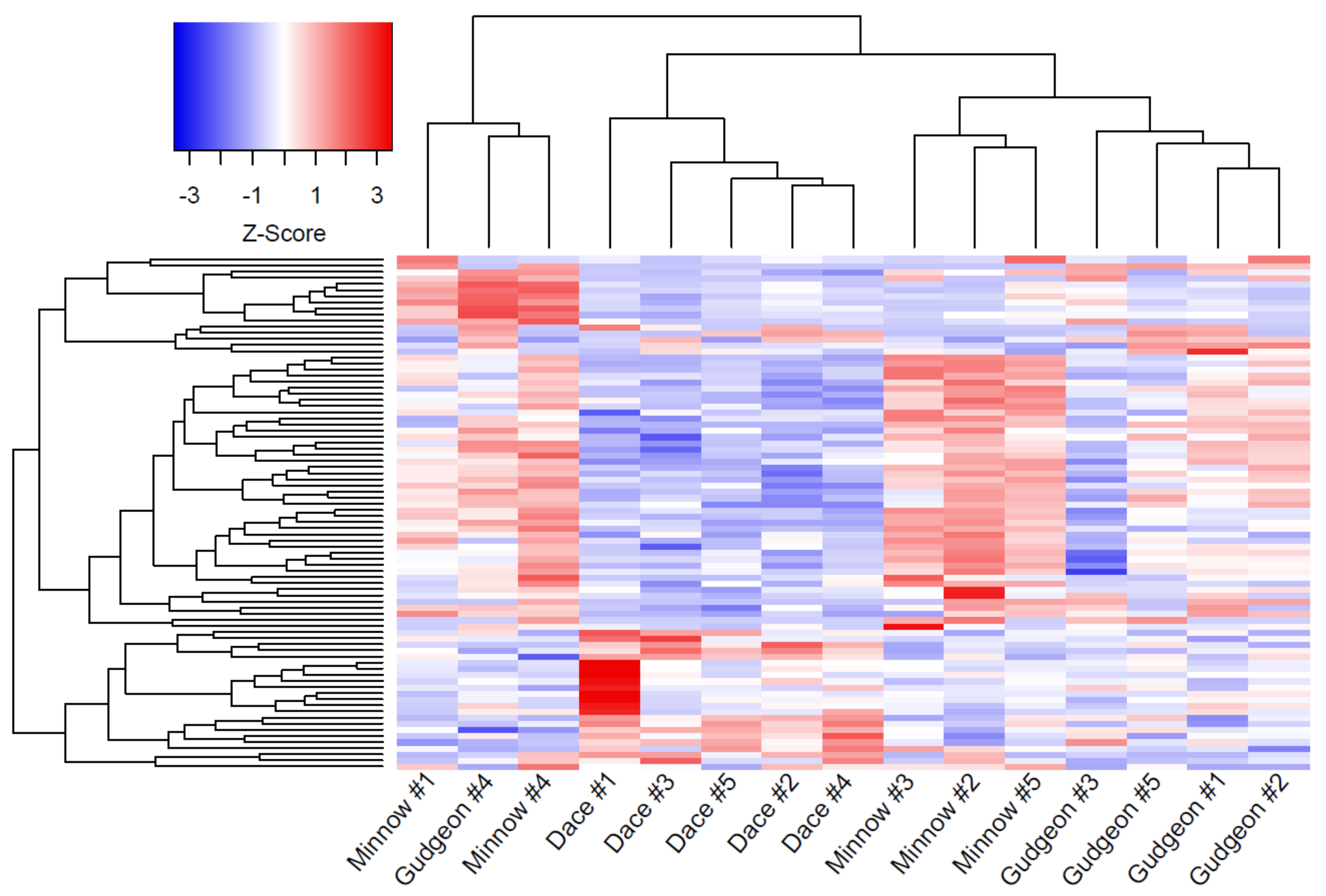
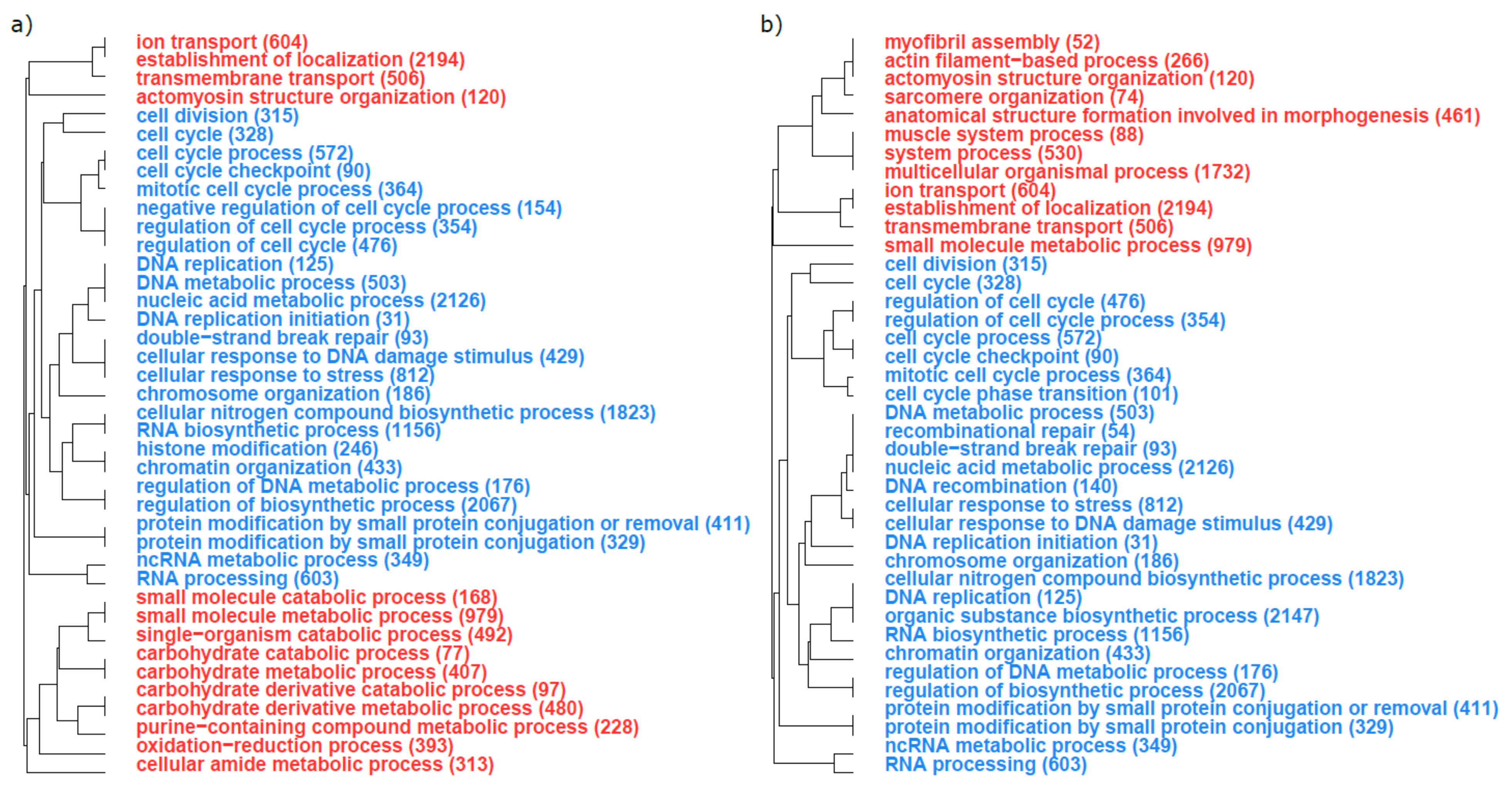
3. Results
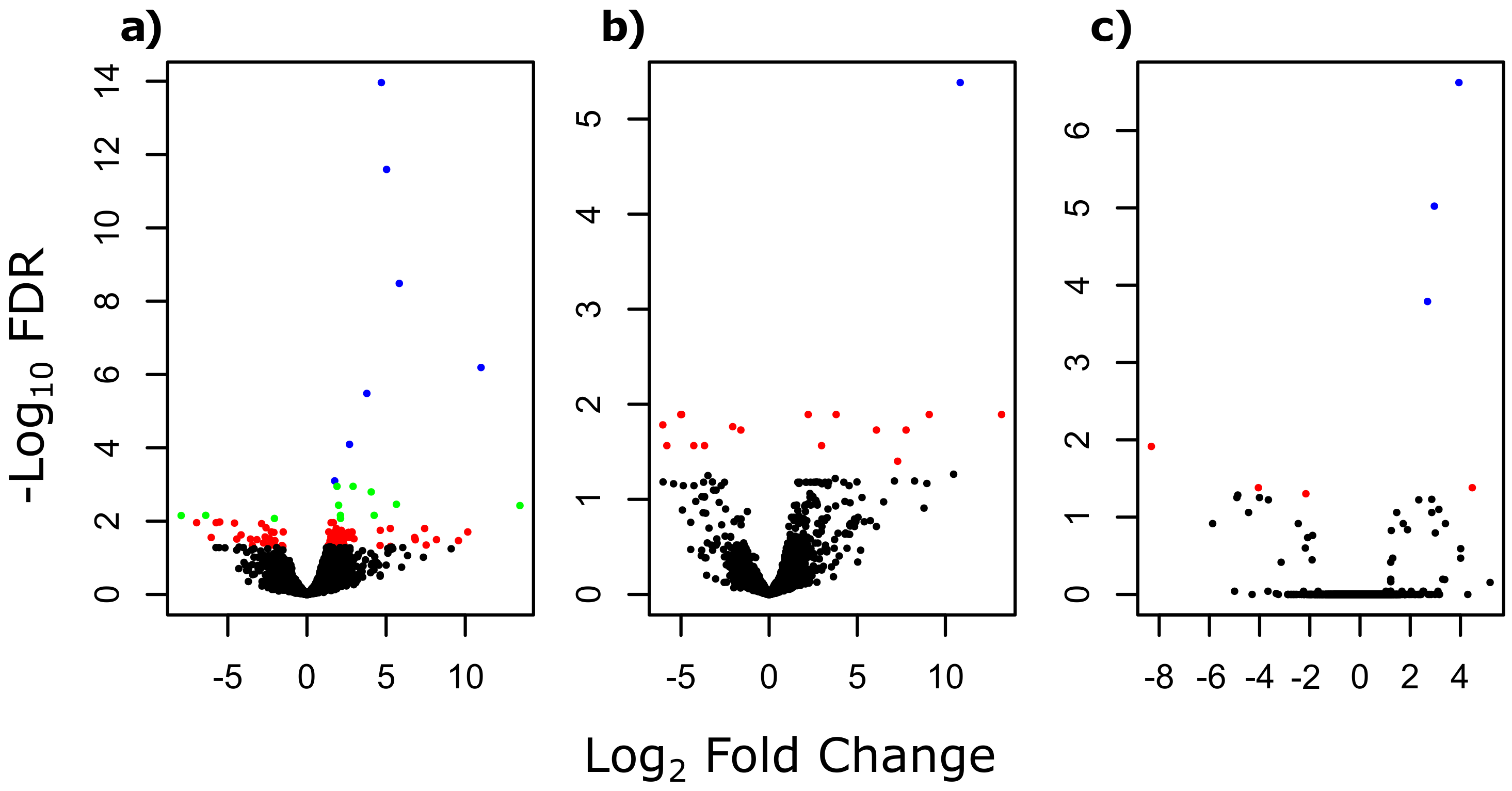
3.1. Differentially Expressed Genes
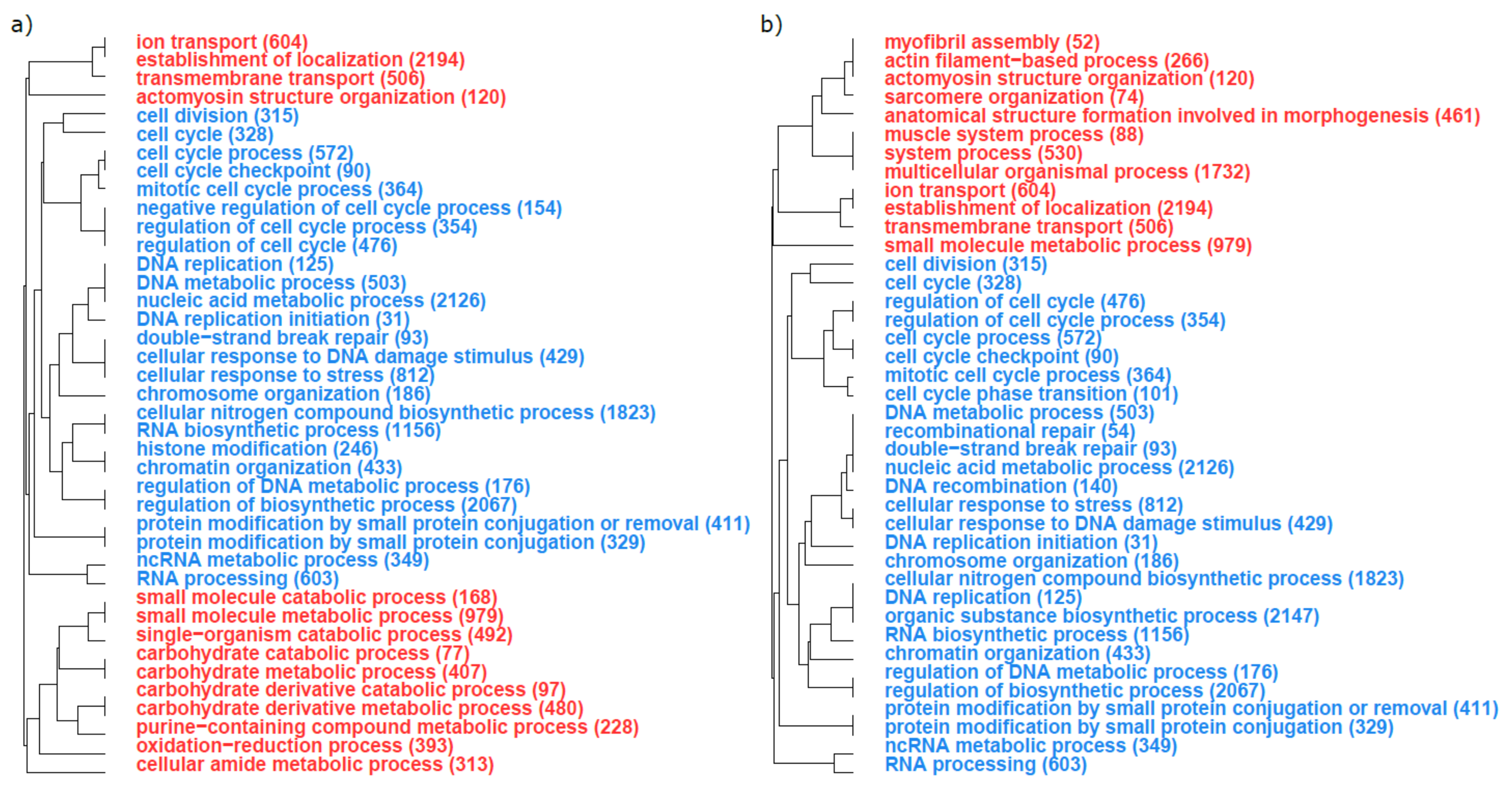
3.2. Functional Analyses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ronquist, F. Parsimony analysis of coevolving species associations. In Tangled Trees Phylogeny Cospeciation Coevolution; University of Chicago Press: Chicago, IL, USA, 2003; pp. 22–64. [Google Scholar]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global Trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef] [PubMed]
- Poulin, R.; Keeney, D.B. Host specificity under molecular and experimental scrutiny. Trends Parasitol. 2008, 24, 24–28. [Google Scholar] [CrossRef]
- Agosta, S.J.; Janz, N.; Brooks, D.R. How specialists can be generalists: Resolving the “parasite paradox” and implications for emerging infectious disease. Zoologia 2010, 27, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Thompson, R.C.A.; Kutz, S.; Smith, A. Parasite zoonoses and wildlife: Emerging issues. Int. J. Environ. Res. Public Health 2009, 6, 678–693. [Google Scholar] [CrossRef]
- Dunn, A.M. Parasites and biological invasions. Adv. Parasitol. 2009, 68, 161–184. [Google Scholar]
- Moir, M.L.; Vesk, P.A.; Brennan, K.E.C.; Keith, D.A.; Hughes, L.; McCarthy, M.A. Current constraints and future directions in estimating coextinction: Constraints and directions. Conserv. Biol. 2010, 24, 682–690. [Google Scholar] [CrossRef]
- Daszak, P. Emerging infectious diseases of wildlife—Threats to biodiversity and human health. Science 2000, 287, 443–449. [Google Scholar] [CrossRef]
- Araujo, S.B.L.; Braga, M.P.; Brooks, D.R.; Agosta, S.J.; Hoberg, E.P.; von Hartenthal, F.W.; Boeger, W.A. Understanding host-switching by ecological fitting. PLoS ONE 2015, 10, e0139225. [Google Scholar] [CrossRef] [Green Version]
- Janzen, D.H. On ecological fitting. Oikos 1985, 45, 308–310. [Google Scholar] [CrossRef] [Green Version]
- De Fine Licht, H.H. Does pathogen plasticity facilitate host shifts? PLoS Pathog. 2018, 14, e1006961. [Google Scholar] [CrossRef] [Green Version]
- Nylin, S.; Agosta, S.; Bensch, S.; Boeger, W.A.; Braga, M.P.; Brooks, D.R.; Forister, M.L.; Hambäck, P.A.; Hoberg, E.P.; Nyman, T.; et al. Embracing colonizations: A new paradigm for species association dynamics. Trends Ecol. Evol. 2018, 33, 4–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lootvoet, A.; Blanchet, S.; Gevrey, M.; Buisson, L.; Tudesque, L.; Loot, G. Patterns and processes of alternative host use in a generalist parasite: Insights from a natural host-parasite interaction. Funct. Ecol. 2013, 27, 1403–1414. [Google Scholar] [CrossRef] [Green Version]
- Little, T.J.; Watt, K.; Ebert, D. Parasite-host specificity: Experimental studies on the basis of parasite adaptation. Evolution 2006, 60, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Mácová, A.; Hoblíková, A.; Hypša, V.; Stanko, M.; Martinů, J.; Kvičerová, J. Mysteries of host switching: Diversification and host specificity in rodent-coccidia associations. Mol. Phylogenetics Evol. 2018, 127, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Rey, O.; Fourtune, L.; Paz-Vinas, I.; Loot, G.; Veyssière, C.; Roche, B.; Blanchet, S. Elucidating the spatio-temporal dynamics of an emerging wildlife pathogen using approximate Bayesian computation. Mol. Ecol. 2015, 24, 5348–5363. [Google Scholar] [CrossRef] [PubMed]
- Mazé-Guilmo, E. Etude du Potentiel de Colonisation des Parasites: Une Approche Intégrative; Université Paul Sabatier (Toulouse III): Toulouse, France, 2016. [Google Scholar]
- Gibbons, T.C.; Metzger, D.C.H.; Healy, T.M.; Schulte, P.M. Gene expression plasticity in response to salinity acclimation in threespine stickleback ecotypes from different salinity habitats. Mol. Ecol. 2017, 26, 2711–2725. [Google Scholar] [CrossRef]
- Fryer, G. The Parasitic Copepoda and Branchiura of British Freshwater Fishes. A Handbook and Key; Freshwater Biological Association: Ambleside, UK, 1982. [Google Scholar]
- von Nordmann, A. Mikrographische Beiträge zur Naturgeschichte der Wirbellosen Thiere; Reimer: Berlin, Germany, 1832; Volume 2. [Google Scholar]
- Monod, T.; Vladykov, V. Sur quelques copépodes parasites provenant de la Russie sous-carpathique (Tchécoslovaquie). Ann. Parasitol. Hum. Comp. 1931, 9, 202–224. [Google Scholar] [CrossRef]
- Loot, G.; Poulet, N.; Reyjol, Y.; Blanchet, S.; Lek, S. The effects of the ectoparasite Tracheliastes polycolpus (copepoda: Lernaeopodidae) on the fins of rostrum dace (Leuciscus leuciscus burdigalensis). Parasitol. Res. 2004, 94, 16–23. [Google Scholar] [CrossRef]
- Blanchet, S.; Méjean, L.; Bourque, J.-F.; Lek, S.; Thomas, F.; Marcogliese, D.J.; Dodson, J.J.; Loot, G. Why do parasitized hosts look different? resolving the “chicken-egg” dilemma. Oecologia 2009, 160, 37–47. [Google Scholar] [CrossRef]
- Mathieu-Bégné, E.; Loot, G.; Blanchet, S.; Toulza, E.; Genthon, C.; Rey, O. De novo transcriptome assembly for Tracheliastes polycolpus, an invasive ectoparasite of freshwater fish in western Europe. Mar. Genom. 2019, 46, 58–61. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2017. [Google Scholar]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [Green Version]
- Storey, J.D. False Discovery Rate. In International Encyclopedia of Statistical Science; Springer: Berlin/Heidelberg, Germany, 2011; pp. 504–508. [Google Scholar]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Wright, R.M.; Aglyamova, G.V.; Meyer, E.; Matz, M.V. Gene expression associated with white syndromes in a reef building coral, Acropora hyacinthus. BMC Genom. 2015, 16, 371. [Google Scholar] [CrossRef] [Green Version]
- Mathieu-Bégné, E.; Loot, G.; Mazé-Guilmo, E.; Mullet, V.; Genthon, C.; Blanchet, S. Combining species distribution models and population genomics underlines the determinants of range limitation in an emerging parasite. Ecography 2021, 44, 307–3019. [Google Scholar] [CrossRef]
- Foll, M.; Gaggiotti, O. A Genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef] [Green Version]
- Poulin, R.; Hamilton, W.J. Egg size variation as a function of environmental variability in parasitic trematodes. Can. J. Zoolog. 2000, 78, 6. [Google Scholar] [CrossRef]
- Loot, G.; Blanchet, S.; Aldana, M.; Navarrete, S.A. Evidence of plasticity in the reproduction of a trematode parasite: The effect of host removal. J. Parasitol. 2008, 94, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.; Reece, S.E.; Drew, D.R.; Haydon, D.T.; Yates, A.J. Plasticity in transmission strategies of the malaria parasite, Plasmodium chabaudi: Environmental and genetic effects. Evol. Appl. 2013, 6, 365–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birget, P.L.G.; Repton, C.; O’Donnell, A.J.; Schneider, P.; Reece, S.E. Phenotypic plasticity in reproductive effort: Malaria parasites respond to resource availability. Proc. R. Soc. B 2017, 284, 20171229. [Google Scholar] [CrossRef] [Green Version]
- Shocket, M.S.; Vergara, D.; Sickbert, A.J.; Walsman, J.M.; Strauss, A.T.; Hite, J.L.; Duffy, M.A.; Cáceres, C.E.; Hall, S.R. Parasite rearing and infection temperatures jointly influence disease transmission and shape seasonality of epidemics. Ecology 2018, 99, 1975–1987. [Google Scholar] [CrossRef] [Green Version]
- Hébert, F.O.; Grambauer, S.; Barber, I.; Landry, C.R.; Aubin-Horth, N. Major host transitions are modulated through transcriptome-wide reprogramming events in Schistocephalus solidus, a threespine stickleback parasite. Mol. Ecol. 2017, 26, 1118–1130. [Google Scholar] [CrossRef] [Green Version]
- Torres-Sánchez, M.; Villate, J.; McGrath-Blaser, S.; Longo, A.V. Panzootic chytrid fungus exploits diverse amphibian host environments through plastic infection strategies. bioRxiv 2021, 11, 470466. [Google Scholar] [CrossRef]
- Storz, G.; Imlayt, J.A. Oxidative stress. Curr. Opin. Microbiol. 1999, 2, 188–194. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathieu-Bégné, E.; Blanchet, S.; Mitta, G.; Le Potier, C.; Loot, G.; Rey, O. Transcriptomic Adjustments in a Freshwater Ectoparasite Reveal the Role of Molecular Plasticity for Parasite Host Shift. Genes 2022, 13, 525. https://doi.org/10.3390/genes13030525
Mathieu-Bégné E, Blanchet S, Mitta G, Le Potier C, Loot G, Rey O. Transcriptomic Adjustments in a Freshwater Ectoparasite Reveal the Role of Molecular Plasticity for Parasite Host Shift. Genes. 2022; 13(3):525. https://doi.org/10.3390/genes13030525
Chicago/Turabian StyleMathieu-Bégné, Eglantine, Simon Blanchet, Guillaume Mitta, Clément Le Potier, Géraldine Loot, and Olivier Rey. 2022. "Transcriptomic Adjustments in a Freshwater Ectoparasite Reveal the Role of Molecular Plasticity for Parasite Host Shift" Genes 13, no. 3: 525. https://doi.org/10.3390/genes13030525
APA StyleMathieu-Bégné, E., Blanchet, S., Mitta, G., Le Potier, C., Loot, G., & Rey, O. (2022). Transcriptomic Adjustments in a Freshwater Ectoparasite Reveal the Role of Molecular Plasticity for Parasite Host Shift. Genes, 13(3), 525. https://doi.org/10.3390/genes13030525