
enChIP-Seq Analyzer: A Software Program to Analyze and Interpret enChIP-Seq Data for the Detection of Physical Interactions between Genomic Regions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Implementation
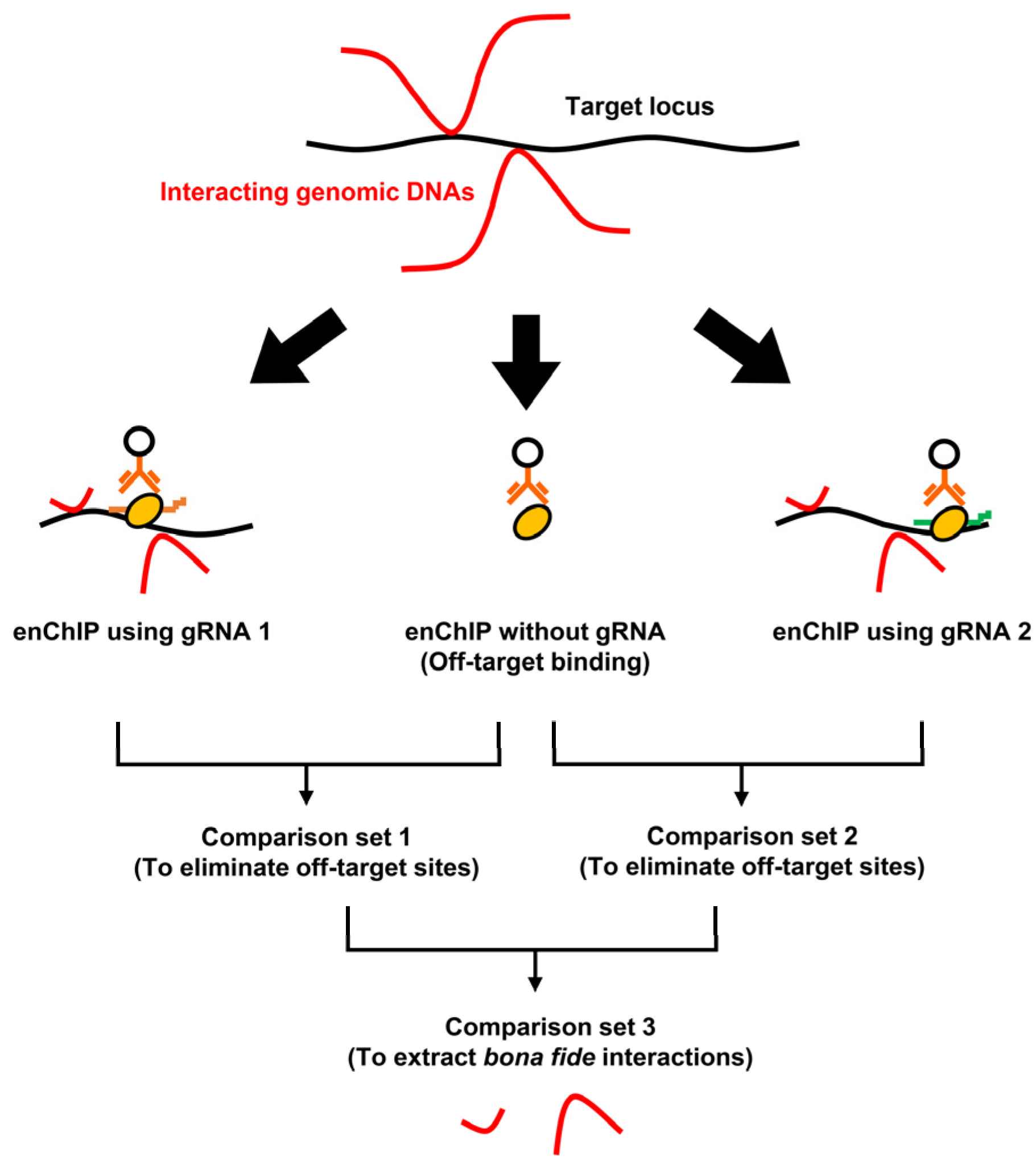
2.2. Procedures for enChIP-Seq
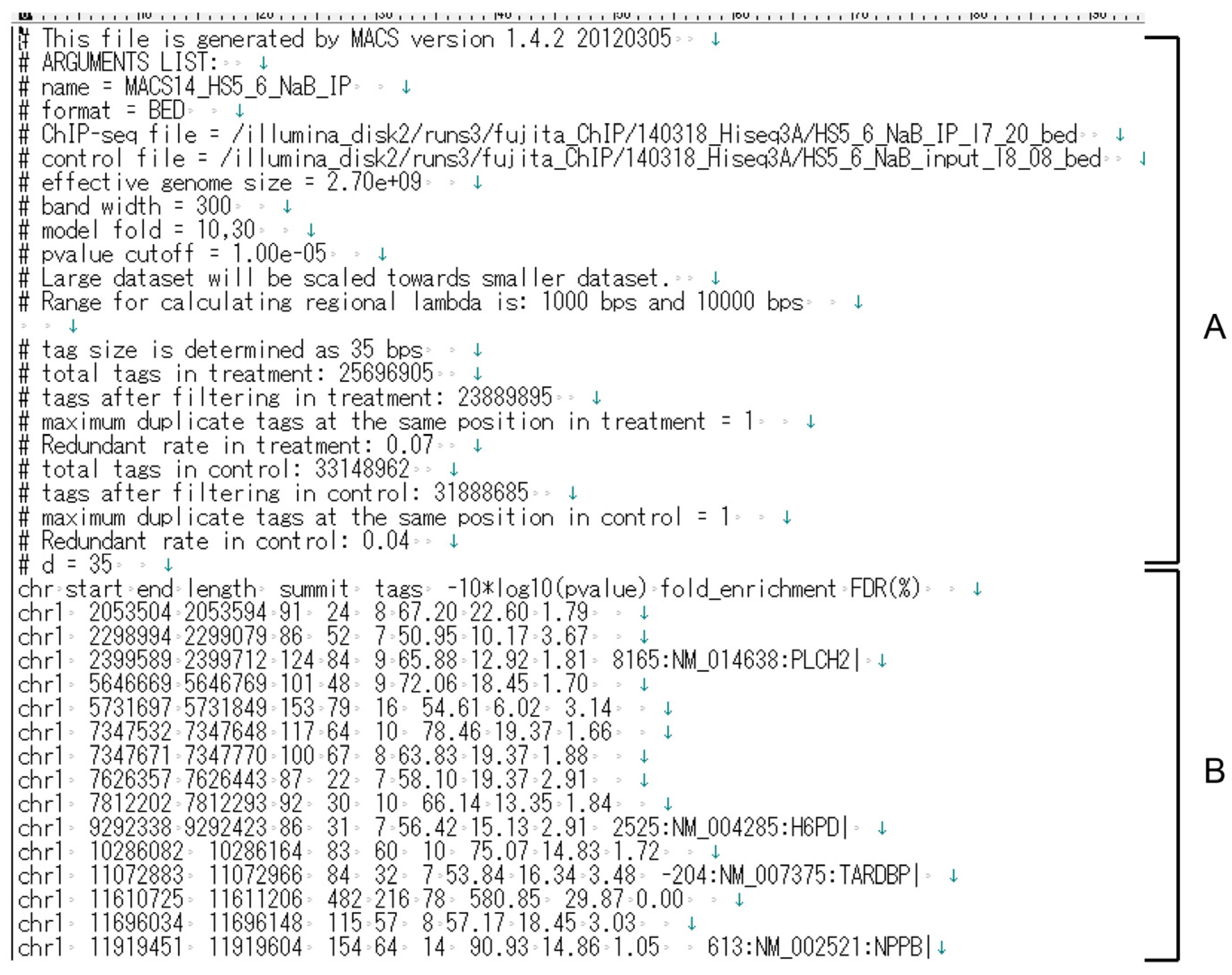
2.3. Filtering of the NGS Peaks to Identify Genomic Regions Interacting with a Target Locus
2.4. Preparation of the Data Set
2.5. Mode of Analysis
2.6. Handling of the System
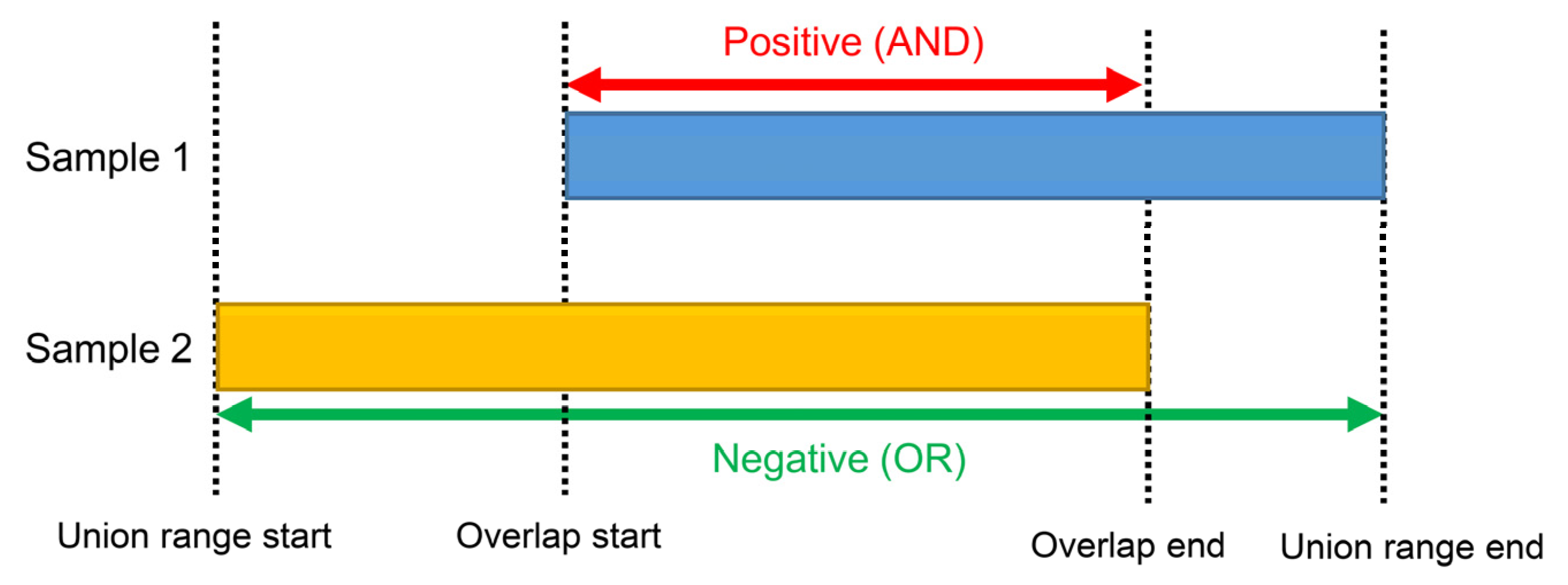
2.6.1. Extraction of Peak Information Commonly Included in Two or More Tab Files
2.6.2. Elimination of Negative Peak Information
2.6.3. Data Export
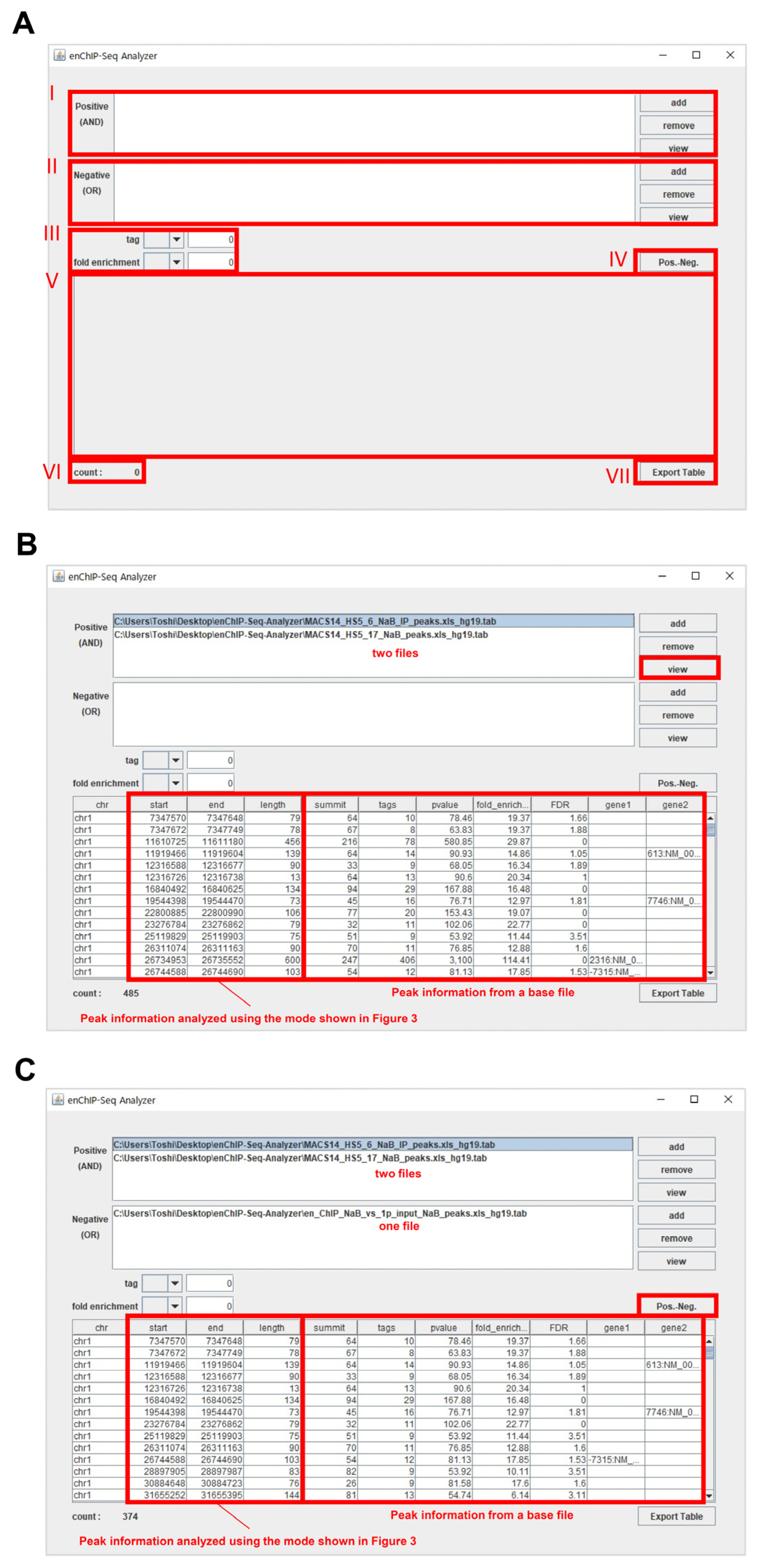
2.6.4. View Information within a Tab File
- (1)
- The information within a tab file can be viewed directly in the software program. To this end, add one tab file that includes enChIP-specific NGS peaks in the field “I” (or “II”).
- (2)
- Click on “view” in the field “I” (or “II”) to view the information within the file.
- (3)
- The information is shown in “V”.
3. Results and Discussion
- Step 1:
- (1)
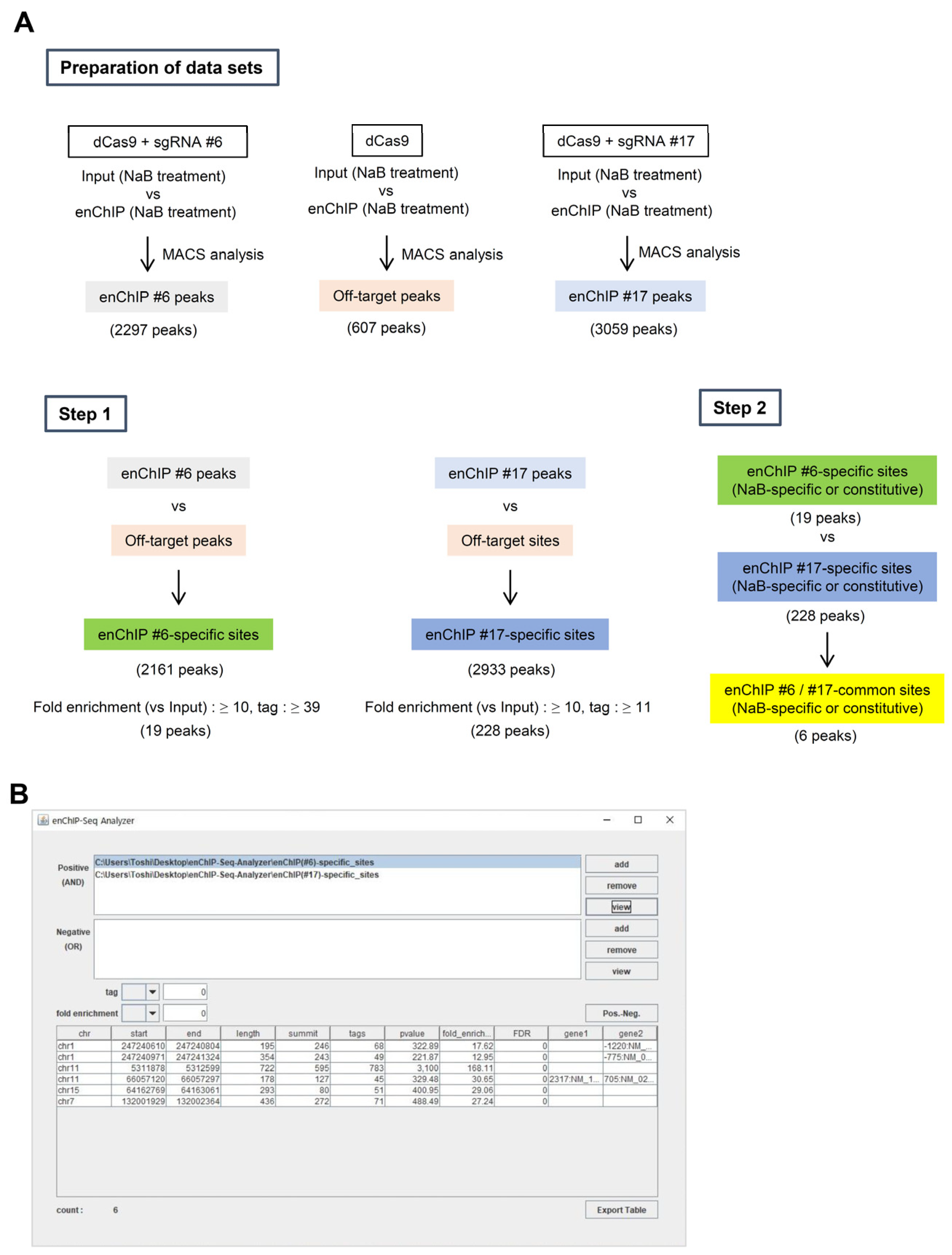
- We compared the data from “enChIP #6 peaks” and “Off-target sites” to eliminate off-target binding sites. The resultant information was named “enChIP #6-specific sites”.
- (2)
- We compared the data from “enChIP #17 peaks” and “Off-target sites” to eliminate off-target binding sites. The resultant information was named “enChIP #17-specific sites”.
- (3)
- To identify peaks with confidence, we adopted two criteria for choosing peaks based on NGS information from the target 5′HS5 locus: (i) Tag number ≥5% of that of the target 5′HS5 locus (which can be considered as an interacting ratio of ≥5%) and (ii) fold enrichment relative to input genomic DNA ≥10. In this regard, 19 and 228 peaks for “enChIP #6-specific sites” and “enChIP #17-specific sites”, respectively, passed the two criteria.
- Step 2:
- We compared the data from “enChIP #6-specific sites” and “enChIP #17-specific sites” to extract “enChIP #6/#17-common sites”. We extracted six peaks, which can be considered bona fide physically interacting genomic regions (Figure 5B). These results were consistent with those from our previous study [10] (please note that in Ref. [10], the symbols > should have been ≥).
4. Conclusions
5. Availability and Requirements
- Project name: enChIP-Seq analyzer
- Software homepage (GitHub): https://github.com/TKY-SE/enChIP-Seq-Analyzer, accessed on 1 December 2021
- Programming language: Java
- Other requirements: Windows machine
- License: None
- Any restrictions to use by non-academics: None.
- Notes: We have a plan to adapt the software for Linux and Mac. As soon as they are available, we will upload the code in GitHub
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Misteli, T. The Self-organizing genome: Principles of genome architecture and function. Cell 2020, 183, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Krumm, A.; Duan, Z. Understanding the 3D genome: Emerging impacts on human disease. Semin. Cell Dev. Biol. 2019, 90, 62–77. [Google Scholar] [CrossRef] [PubMed]
- Dekker, J.; Rippe, K.; Dekker, M.; Kleckner, N. Capturing chromosome conformation. Science 2002, 295, 1306–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Wit, E.; de Laat, W. A decade of 3C technologies: Insights into nuclear organization. Genes Dev. 2012, 26, 11–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, I.; Berlivet, S.; Eskeland, R.; Boyle, S.; Illingworth, R.S.; Paquette, D.; Dostie, J.; Bickmore, W.A. Spatial genome organization: Contrasting views from chromosome conformation capture and fluorescence in situ hybridization. Genes Dev. 2014, 28, 2778–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, E.H.; Pegoraro, G.; Brandão, H.B.; Valton, A.L.; Oomen, M.E.; Dekker, J.; Mirny, L.; Misteli, T. Extensive heterogeneity and intrinsic variation in spatial genome organization. Cell 2019, 176, 1502–1515.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beagrie, R.A.; Scialdone, A.; Schueler, M.; Kraemer, D.C.A.; Chotalia, M.; Xie, S.Q.; Barbieri, M.; de Santiago, I.; Lavitas, L.M.; Branco, M.R.; et al. Complex multi-enhancer contacts captured by genome architecture mapping. Nature 2017, 543, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Quinodoz, S.A.; Ollikainen, N.; Tabak, B.; Palla, A.; Schmidt, J.M.; Detmar, E.; Lai, M.M.; Shishkin, A.A.; Bhat, P.; Takei, Y.; et al. Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell 2018, 174, 744–757.e24. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Tian, S.Z.; Capurso, D.; Kim, M.; Maurya, R.; Lee, B.; Piecuch, E.; Gong, L.; Zhu, J.J.; Li, Z.; et al. Multiplex chromatin interactions with single-molecule precision. Nature 2019, 566, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Yuno, M.; Suzuki, Y.; Sugano, S.; Fujii, H. Identification of physical interactions between genomic regions by enChIP-Seq. Genes Cells 2017, 22, 506–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, T.; Kitaura, F.; Yuno, M.; Suzuki, Y.; Sugano, S.; Fujii, H. Locus-specific ChIP combined with NGS analysis reveals genomic regulatory regions that physically interact with the Pax5 promoter in a chicken B cell line. DNA Res. 2017, 24, 537–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cencic, R.; Miura, H.; Malina, A.; Robert, F.; Ethier, S.; Schmeing, T.M.; Dostie, J.; Pelletier, J. Protospacer adjacent motif (PAM)-distal sequences engage CRISPR Cas9 DNA target cleavage. PLoS ONE 2014, 9, e109213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuscu, C.; Arslan, S.; Singh, R.; Thorpe, J.; Adli, M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 2014, 32, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Scott, D.A.; Kriz, A.J.; Chiu, A.C.; Hsu, P.D.; Dadon, D.B.; Cheng, A.W.; Trevino, A.E.; Konermann, S.; Chen, S.; et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 2014, 32, 670–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Geen, H.; Henry, I.M.; Bhakta, M.S.; Meckler, J.F.; Segal, D.J. A genome-wide analysis of Cas9 binding specificity using ChIP-seq and targeted sequence capture. Nucleic Acids Res. 2015, 43, 3389–3404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarudate, A.; Fujita, T.; Nakayama, T.; Fujii, H. enChIP-Seq Analyzer: A Software Program to Analyze and Interpret enChIP-Seq Data for the Detection of Physical Interactions between Genomic Regions. Genes 2022, 13, 472. https://doi.org/10.3390/genes13030472
Sarudate A, Fujita T, Nakayama T, Fujii H. enChIP-Seq Analyzer: A Software Program to Analyze and Interpret enChIP-Seq Data for the Detection of Physical Interactions between Genomic Regions. Genes. 2022; 13(3):472. https://doi.org/10.3390/genes13030472
Chicago/Turabian StyleSarudate, Ashita, Toshitsugu Fujita, Takahiro Nakayama, and Hodaka Fujii. 2022. "enChIP-Seq Analyzer: A Software Program to Analyze and Interpret enChIP-Seq Data for the Detection of Physical Interactions between Genomic Regions" Genes 13, no. 3: 472. https://doi.org/10.3390/genes13030472
APA StyleSarudate, A., Fujita, T., Nakayama, T., & Fujii, H. (2022). enChIP-Seq Analyzer: A Software Program to Analyze and Interpret enChIP-Seq Data for the Detection of Physical Interactions between Genomic Regions. Genes, 13(3), 472. https://doi.org/10.3390/genes13030472