
SINE-Based Phylogenomics Reveal Extensive Introgression and Incomplete Lineage Sorting in Myotis


Abstract
1. Introduction
2. Materials and Methods
3. Results
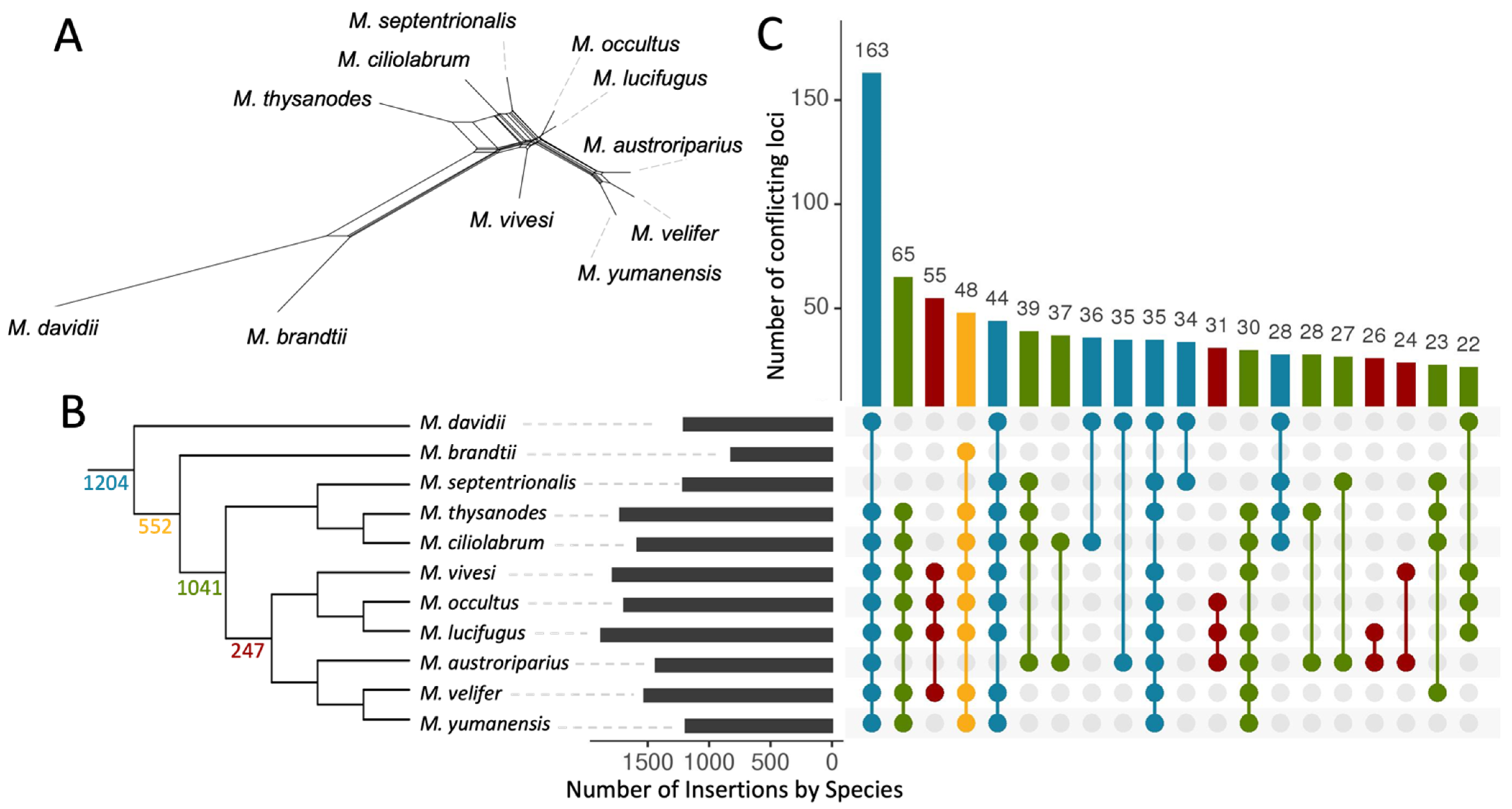
3.1. WGS Mapping and TE Variation Discovery
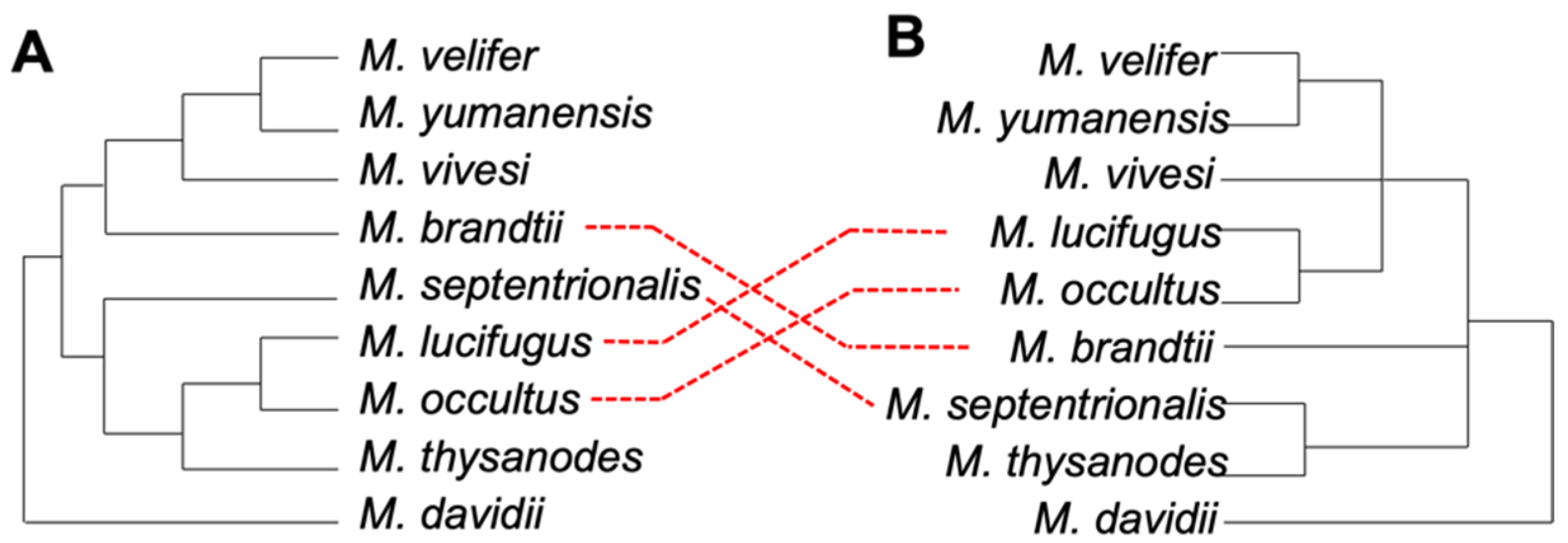
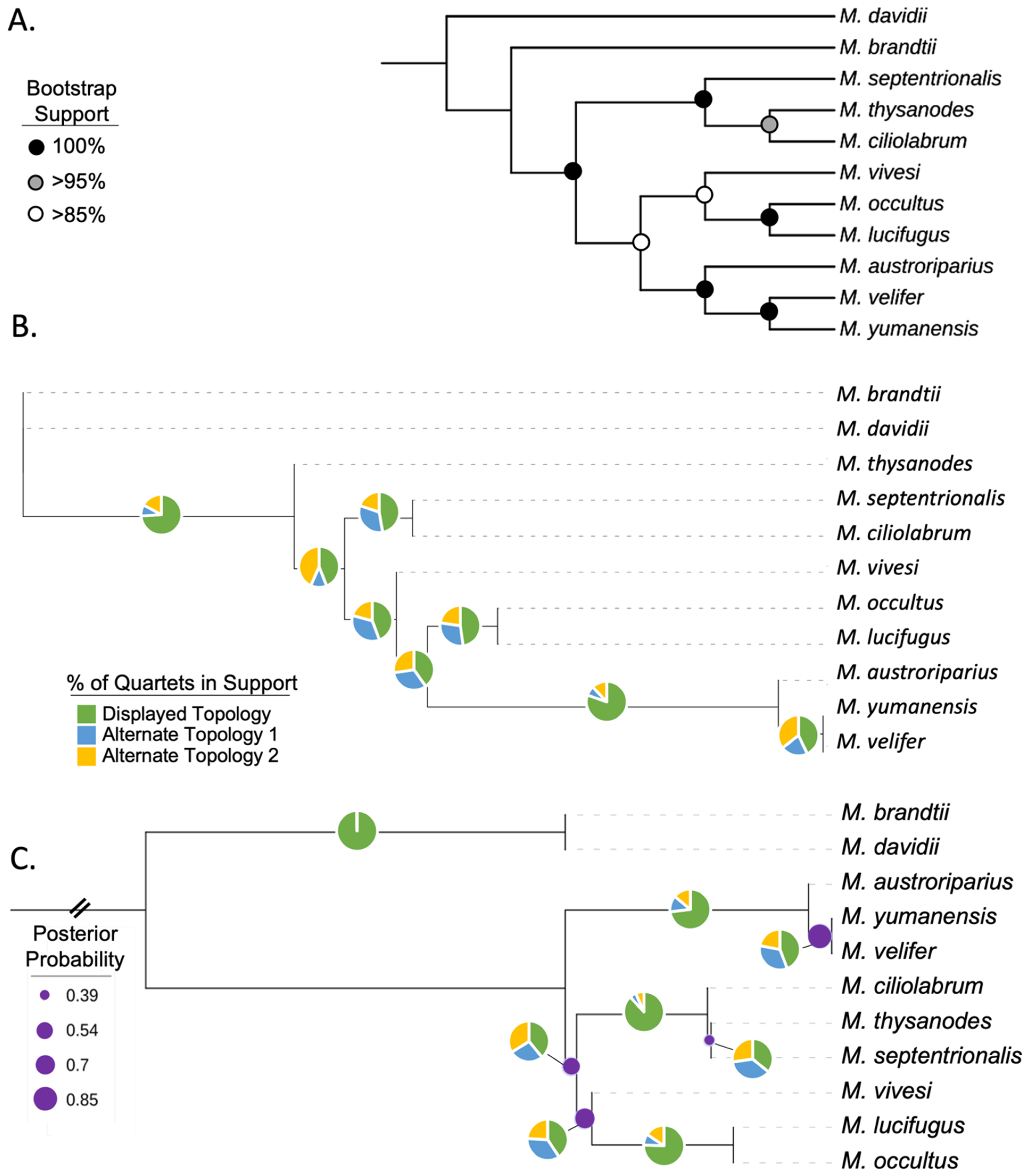
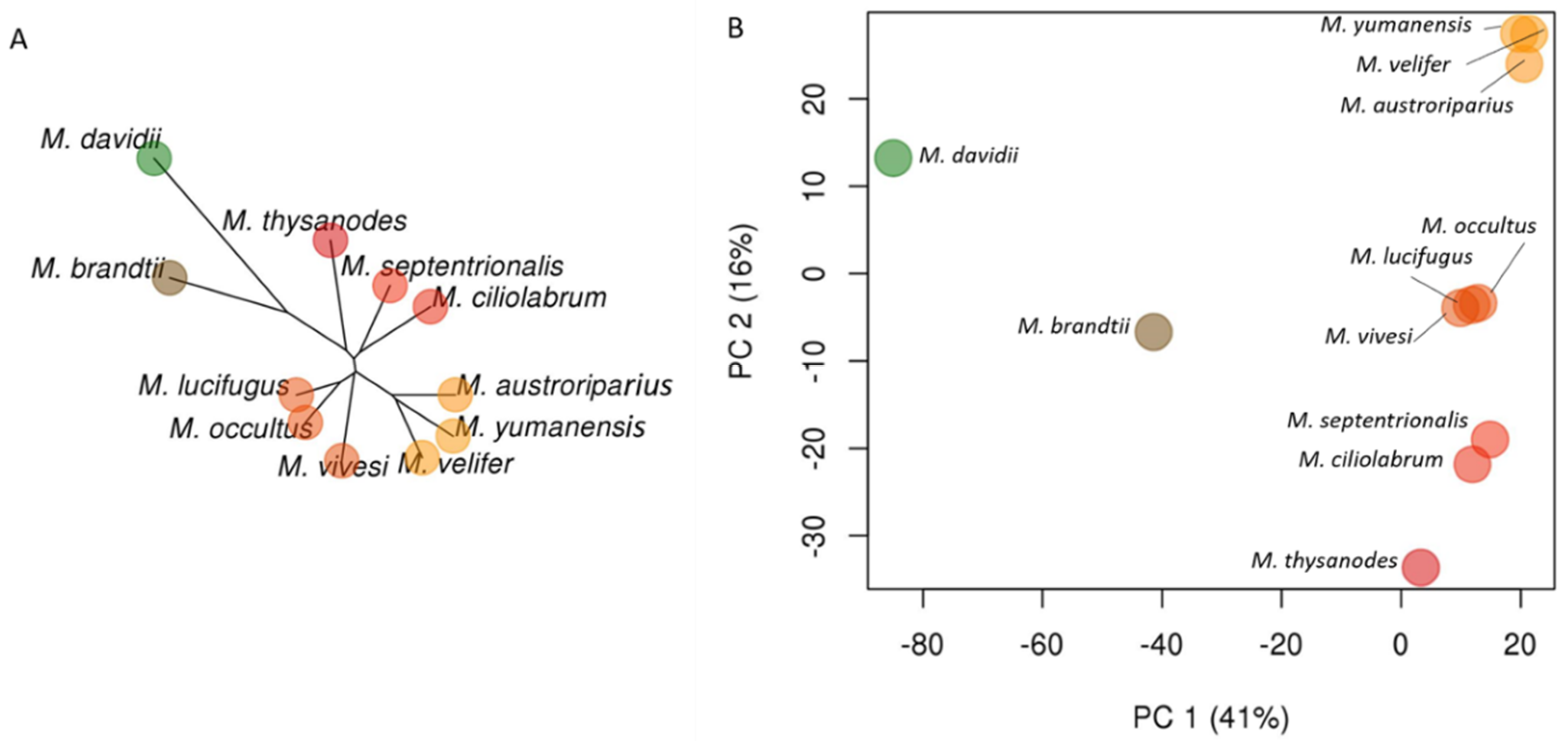
3.2. Phylogenetic Relationships in Myotis
3.3. Ves Insertion Discordance
3.4. UCE Phylogeny
3.5. Evidence of Gene Flow
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Churakov, G.; Kriegs, J.O.; Baertsch, R.; Zemann, A.; Brosius, J.; Schmitz, J. Mosaic Retroposon Insertion Patterns in Placental Mammals. Genome Res. 2009, 19, 868–887. [Google Scholar] [CrossRef] [PubMed]
- Kriegs, J.O.; Churakov, G.; Kiefmann, M.; Jordan, U.; Brosius, J.; Schmitz, J. Retroposed Elements as Archives for the Evolutionary History of Placental Mammals. PLoS Biol. 2006, 4, e91. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, H.; Maruyama, S.; Okada, N. Retroposon Analysis and Recent Geological Data Suggest Near-Simultaneous Divergence of the Three Superorders of Mammals. Proc. Natl. Acad. Sci. USA 2009, 106, 5235–5240. [Google Scholar] [CrossRef] [PubMed]
- Lammers, F.; Blumer, M.; Rücklé, C.; Nilsson, M.A. Retrophylogenomics in Rorquals Indicate Large Ancestral Population Sizes and a Rapid Radiation. Mob. DNA 2019, 10, 5. [Google Scholar] [CrossRef]
- Lavoie, C.A.; Platt, R.N.; Novick, P.A.; Counterman, B.A.; Ray, D.A. Transposable Element Evolution in Heliconius Suggests Genome Diversity within Lepidoptera. Mob. DNA 2013, 4, 21. [Google Scholar] [CrossRef]
- Ray, D.A.; Xing, J.; Salem, A.H.; Batzer, M.A. SINEs of a Nearly Perfect Character. Syst. Biol. 2006, 55, 928–935. [Google Scholar] [CrossRef]
- Doronina, L.; Reising, O.; Clawson, H.; Ray, D.A.; Schmitz, J. True Homoplasy of Retrotransposon Insertions in Primates. Syst. Biol. 2019, 68, 482–493. [Google Scholar] [CrossRef]
- Suh, A.; Smeds, L.; Ellegren, H. The Dynamics of Incomplete Lineage Sorting across the Ancient Adaptive Radiation of Neoavian Birds. PLoS Biol. 2015, 13, e1002224. [Google Scholar] [CrossRef]
- Agnarsson, I.; Zambrana-Torrelio, C.M.; Flores-Saldana, N.P.; May-Collado, L.J. A Time-Calibrated Species-Level Phylogeny of Bats (Chiroptera, Mammalia). PLoS Curr. 2011, 3, RRN1212. [Google Scholar] [CrossRef]
- Stadelmann, B.; Herrera, L.G.; Arroyo-Cabrales, J.; Flores-Martínez, J.J.; May, B.P.; Ruedi, M. Molecular Systematics of the Fishing Bat Myotis (Pizonyx) Vivesi. J. Mammal. 2004, 85, 133–139. [Google Scholar] [CrossRef][Green Version]
- Stadelmann, B.; Lin, L.K.; Kunz, T.H.; Ruedi, M. Molecular Phylogeny of New World Myotis (Chiroptera, Vespertilionidae) Inferred from Mitochondrial and Nuclear DNA Genes. Mol. Phylogenet. Evol. 2007, 43, 32–48. [Google Scholar] [CrossRef]
- Ruedi, M.; Stadelmann, B.; Gager, Y.; Douzery, E.J.P.; Francis, C.M.; Lin, L.K.; Guillén-Servent, A.; Cibois, A. Molecular Phylogenetic Reconstructions Identify East Asia as the Cradle for the Evolution of the Cosmopolitan Genus Myotis (Mammalia, Chiroptera). Mol. Phylogenet. Evol. 2013, 69, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Moratelli, R.; Dewynter, M.; Delaval, M.; Catzeflis, F.; Ruedi, M. First Record of Myotis Albescens (Chiroptera, Vespertilionidae) in French Guiana. Biodivers. Data J. 2015, 3, e5314. [Google Scholar] [CrossRef]
- Ruedi, M.; Mayer, F. Molecular Systematics of Bats of the Genus Myotis (Vespertilionidae) Suggests Deterministic Ecomorphological Convergences. Mol. Phylogenet. Evol. 2001, 21, 436–448. [Google Scholar] [CrossRef]
- Platt, R.N.; Faircloth, B.C.; Sullivan, K.A.M.; Kieran, T.J.; Glenn, T.C.; Vandewege, M.W.; Lee, T.E.; Baker, R.J.; Stevens, R.D.; Ray, D.A. Conflicting Evolutionary Histories of the Mitochondrial and Nuclear Genomes in New World Myotis Bats. Syst. Biol. 2018, 67, 236–249. [Google Scholar] [CrossRef]
- Simmons, N.; Cirranello, A. Bat Species of the World: A Taxonomic and Geographic Database. Available online: https://www.batnames.org (accessed on 16 December 2021).
- Morales, A.E.; Carstens, B.C. Evidence That Myotis Lucifugus “Subspecies” Are Five Nonsister Species, despite Gene Flow. Syst. Biol. 2018, 67, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Afonso, E.; Goydadin, A.-C.; Giraudoux, P.; Farny, G. Investigating Hybridization between the Two Sibling Bat Species Myotis Myotis and M. Blythii from Guano in a Natural Mixed Maternity Colony. PLoS ONE 2017, 12, e0170534. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, A. Morphologic Variation and Hybridization in Myotis Yumanensis Sociabilis and Myotis Lucifugus Carissima. J. Mammal. 1979, 60, 489–504. [Google Scholar] [CrossRef]
- Bachanek, J.; Postawa, T. Morphological Evidence for Hybridization in the Sister Species Myotis Myotis and Myotis Oxygnathus (Chiroptera: Vespertilionidae) in the Carpathian Basin. Acta Chiropterologica 2010, 12, 439–448. [Google Scholar] [CrossRef]
- Zhang, G.; Cowled, C.; Shi, Z.; Huang, Z.; Bishop-Lilly, K.A.; Fang, X.; Wynne, J.W.; Xiong, Z.; Baker, M.L.; Zhao, W.; et al. Comparative Analysis of Bat Genomes Provides Insight into the Evolution of Flight and Immunity. Science 2013, 339, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Seim, I.; Fang, X.; Xiong, Z.; Lobanov, A.V.; Huang, Z.; Ma, S.; Feng, Y.; Turanov, A.A.; Zhu, Y.; Lenz, T.L.; et al. Genome Analysis Reveals Insights into Physiology and Longevity of the Brandt’s Bat Myotis Brandtii. Nat. Commun. 2013, 4, 2212. [Google Scholar] [CrossRef] [PubMed]
- Valdez, E.W.; Choate, J.R.; Bogan, M.A.; Yates, T.L. Taxonomic Status of Myotis Occultus. J. Mammal. 1999, 80, 545–552. [Google Scholar] [CrossRef]
- Piaggio, A.J.; Valdez, E.W.; Bogan, M.A.; Spicer, G.S. Systematics of Myotis Occultus (Chiroptera: Vespertilionidae) Inferred from Sequences of Two Mitochondrial Genes. J. Mammal. 2002, 83, 386–395. [Google Scholar] [CrossRef][Green Version]
- Simmons, N. Mammal Species of the World: A Taxonomic and Geographic Reference. In Mammal Species of the World: A Taxonomic and Geographic Reference; Wilson, D., Reader, D., Eds.; Johns Hopkins University Press: Baltimore, MD, USA, 2005; pp. 312–529. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Lindblad-Toh, K.; Garber, M.; Zuk, O.; Lin, M.F.; Parker, B.J.; Washietl, S.; Kheradpour, P.; Ernst, J.; Jordan, G.; Mauceli, E.; et al. A High-Resolution Map of Human Evolutionary Constraint Using 29 Mammals. Nature 2011, 478, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Broad Institute Picard Toolkit. 2019. Available online: http://broadinstitute.github.io/picard/ (accessed on 16 December 2021).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. Subgroup, 1000 Genome Project Data Processing the Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Gardner, E.J.; Lam, V.K.; Harris, D.N.; Chuang, N.T.; Scott, E.C.; Stephen Pittard, W.; Mills, R.E.; Devine, S.E. The Mobile Element Locator Tool (MELT): Population-Scale Mobile Element Discovery and Biology. Genome Res. 2017, 27, 1916–1929. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.A.; Pagan, H.J.T.; Platt, R.N.; Kroll, A.R.; Schaack, S.; Stevens, R.D. Differential SINE Evolution in Vesper and Non-Vesper Bats. Mob. DNA 2015, 6, 10. [Google Scholar] [CrossRef]
- Ray, D.A.; Feschotte, C.; Pagan, H.J.T.; Smith, J.D.; Pritham, E.J.; Arensburger, P.; Atkinson, P.W.; Craig, N.L. Multiple Waves of Recent DNA Transposon Activity in the Bat, Myotis Lucifugus. Genome Res. 2008, 18, 717–728. [Google Scholar] [CrossRef]
- Smit, A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2015. Available online: http://repeatmasker.org/faq.html (accessed on 16 December 2021).
- RM2Bed.Py. Available online: https://github.com/rmhubley/RepeatMasker/blob/master/util/RM2Bed.py (accessed on 21 August 2021).
- SimSeq. Available online: https://github.com/jstjohn/SimSeq (accessed on 21 August 2021).
- Springer, M.S.; Molloy, E.K.; Sloan, D.B.; Simmons, M.P.; Gatesy, J. ILS-Aware Analysis of Low-Homoplasy Retroelement Insertions: Inference of Species Trees and Introgression Using Quartets. J. Hered. 2020, 111, 147–168. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. PHYLIP (Phylogeny Inference Package). 2005. Available online: https://evolution.genetics.washington.edu/phylip.html (accessed on 16 December 2021).
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Zhang, C.; Mirarab, S. ASTRAL-MP: Scaling ASTRAL to Very Large Datasets Using Randomization and Parallelization. Bioinformatics 2019, 35, 3961–3969. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T.; Ahmed, I. Adegenet 1.3-1: New Tools for the Analysis of Genome-Wide SNP Data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R Package for the Visualization of Intersecting Sets and Their Properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4.0b10; Sinauer: Sunderland, MA, USA, 2002. [Google Scholar]
- Holm, S. A Simple Sequentially Rejective Multiple Test Procedure. Scandanavian J. Stat. 1979, 65–70. [Google Scholar]
- Gaetano, J. Holm-Bonferroni Sequential Correction: An Excel Calculator (1.3); Microsoft Excel: Redmond, WA, USA, 2018. [Google Scholar] [CrossRef]
- Faircloth, B.C. PHYLUCE Is a Software Package for the Analysis of Conserved Genomic Loci. Bioinformatics 2016, 32, 786–788. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A Fast, Scalable and User-Friendly Tool for Maximum Likelihood Phylogenetic Inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef]
- Zhang, C.; Rabiee, M.; Sayyari, E.; Mirarab, S. ASTRAL-III: Polynomial Time Species Tree Reconstruction from Partially Resolved Gene Trees. BMC Bioinform. 2018, 19, 153. [Google Scholar] [CrossRef] [PubMed]
- Sayyari, E.; Mirarab, S. Testing for Polytomies in Phylogenetic Species Trees Using Quartet Frequencies. Genes 2018, 9, 132. [Google Scholar] [CrossRef] [PubMed]
- Schwenk, K.; Brede, N.; Streit, B. Introduction. Extent, Processes and Evolutionary Impact of Interspecific Hybridization in Animals. Philos. Trans. R. Soc. B 2008, 363, 2805–2811. [Google Scholar] [CrossRef] [PubMed]
- Molloy, E.K.; Gatesy, J.; Springer, M.S. Theoretical and Practical Considerations when using Retroelement Insertions to Estimate Species Trees in the Anomaly Zone. Syst. Biol. 2021. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | ID Number | Cov. | Accession | Calls | Heterozygosity |
|---|---|---|---|---|---|
| M. austroriparius | M8132 | 19.2x | SRR17555657 | 21,916 | 0.0038 |
| M. brandtii | wgs * | 16x | ANKR00000000 | 17,315 | 0.0004 |
| M. ciliolabrum | TK186048 | 21.8x | SRR17555656 | 21,907 | 0.0043 |
| M. davidii | wgs * | 20.2x | ALWT00000000 | 18,273 | 0.0005 |
| M. occultus | TK186204 | 22.9x | SRR17555655 | 22,220 | 0.0027 |
| M. septentrionalis | RDS8624 | 14.4x | SRR17555654 | 22,098 | 0.0046 |
| M. thysanodes | TK186219 | 24.0x | SRR17555653 | 21,840 | 0.0038 |
| M. velifer | TK167846 | 31.9x | SRR17555652 | 22,070 | 0.0048 |
| M. vivesi | NK5109 | 22.2x | SRR17555651 | 21,892 | 0.0039 |
| M. yumanensis | TK186200 | 19.7x | SRR17555650 | 21,953 | 0.0041 |
| Introgression Pathway | Pathway Number | Quartets Supporting |
|---|---|---|
| M. davidii ↔ ancestor a | I | 34 |
| M. brandtii ↔ ancestor c | II | 55 |
| ancestor b ↔ ancestor c | III | 28 |
| M. brandtii ↔ ancestor b | IV | 9 |
| M. austroriparius ↔ ancestor d | V | 8 |
| M. austroriparius ↔ M. septentrionalis | VI | 12 |
| M. lucifugus ↔ M. septentrionalis | VII | 9 |
| M. thysanodes ↔ M. ciliolabrum | VIII | 7 |
| M. austroriparius ↔ M. lucifigus | VIIII | 13 |
| M. austroriparius ↔ M. velifer | X | 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korstian, J.M.; Paulat, N.S.; Platt, R.N., II; Stevens, R.D.; Ray, D.A. SINE-Based Phylogenomics Reveal Extensive Introgression and Incomplete Lineage Sorting in Myotis. Genes 2022, 13, 399. https://doi.org/10.3390/genes13030399
Korstian JM, Paulat NS, Platt RN II, Stevens RD, Ray DA. SINE-Based Phylogenomics Reveal Extensive Introgression and Incomplete Lineage Sorting in Myotis. Genes. 2022; 13(3):399. https://doi.org/10.3390/genes13030399
Chicago/Turabian StyleKorstian, Jennifer M., Nicole S. Paulat, Roy N. Platt, II, Richard D. Stevens, and David A. Ray. 2022. "SINE-Based Phylogenomics Reveal Extensive Introgression and Incomplete Lineage Sorting in Myotis" Genes 13, no. 3: 399. https://doi.org/10.3390/genes13030399
APA StyleKorstian, J. M., Paulat, N. S., Platt, R. N., II, Stevens, R. D., & Ray, D. A. (2022). SINE-Based Phylogenomics Reveal Extensive Introgression and Incomplete Lineage Sorting in Myotis. Genes, 13(3), 399. https://doi.org/10.3390/genes13030399