
Further Mining and Characterization of miRNA Resource in Chinese Fir (Cunninghamia lanceolata)

 , and
, and 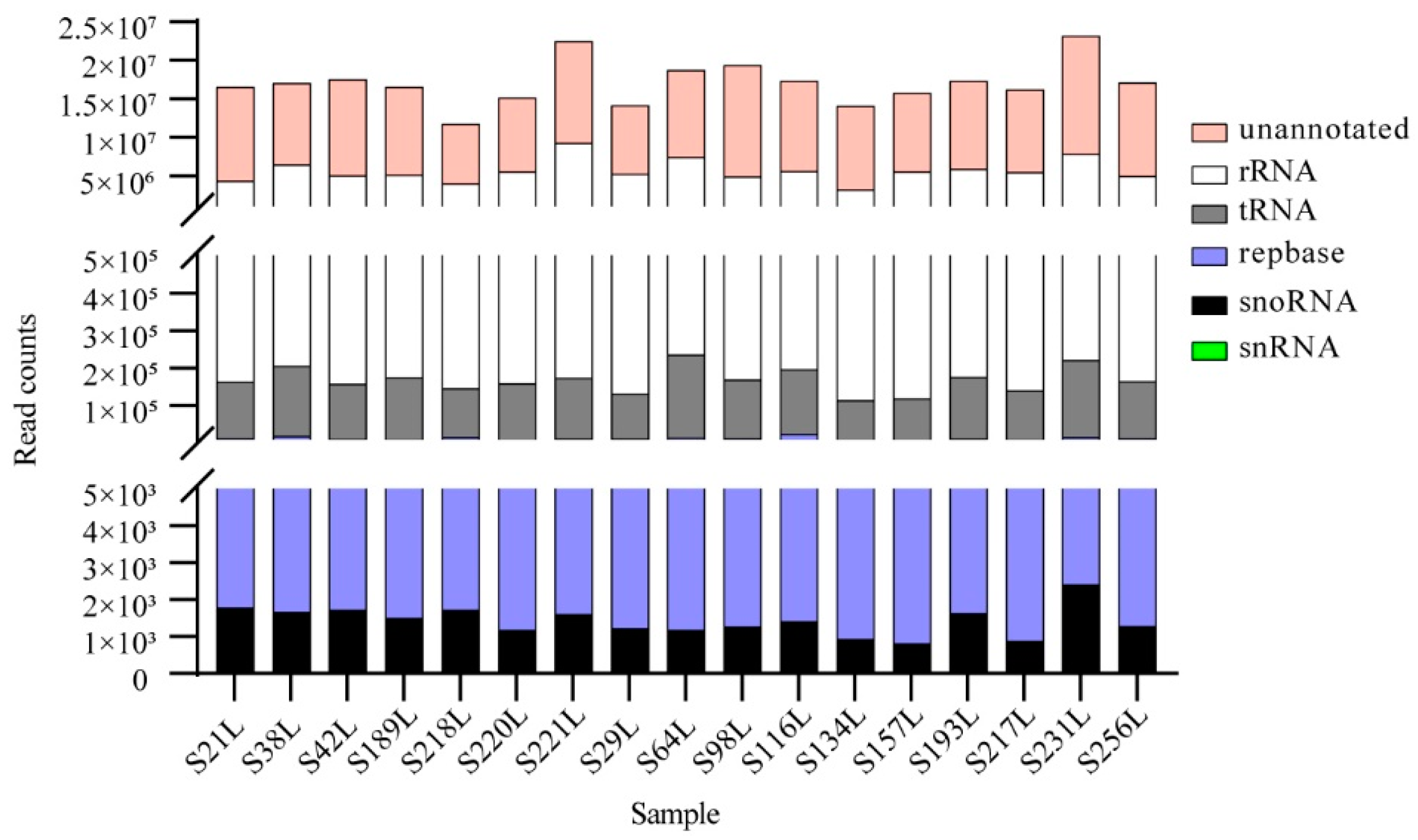{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Extraction, RNA Library Construction and Functional Annotation
2.3. Identification of miRNAs and Prediction of Their Target Gene
2.4. Bioinformatics Analysis
3. Results
3.1. An Overview of High-Throughput Sequencing Datasets
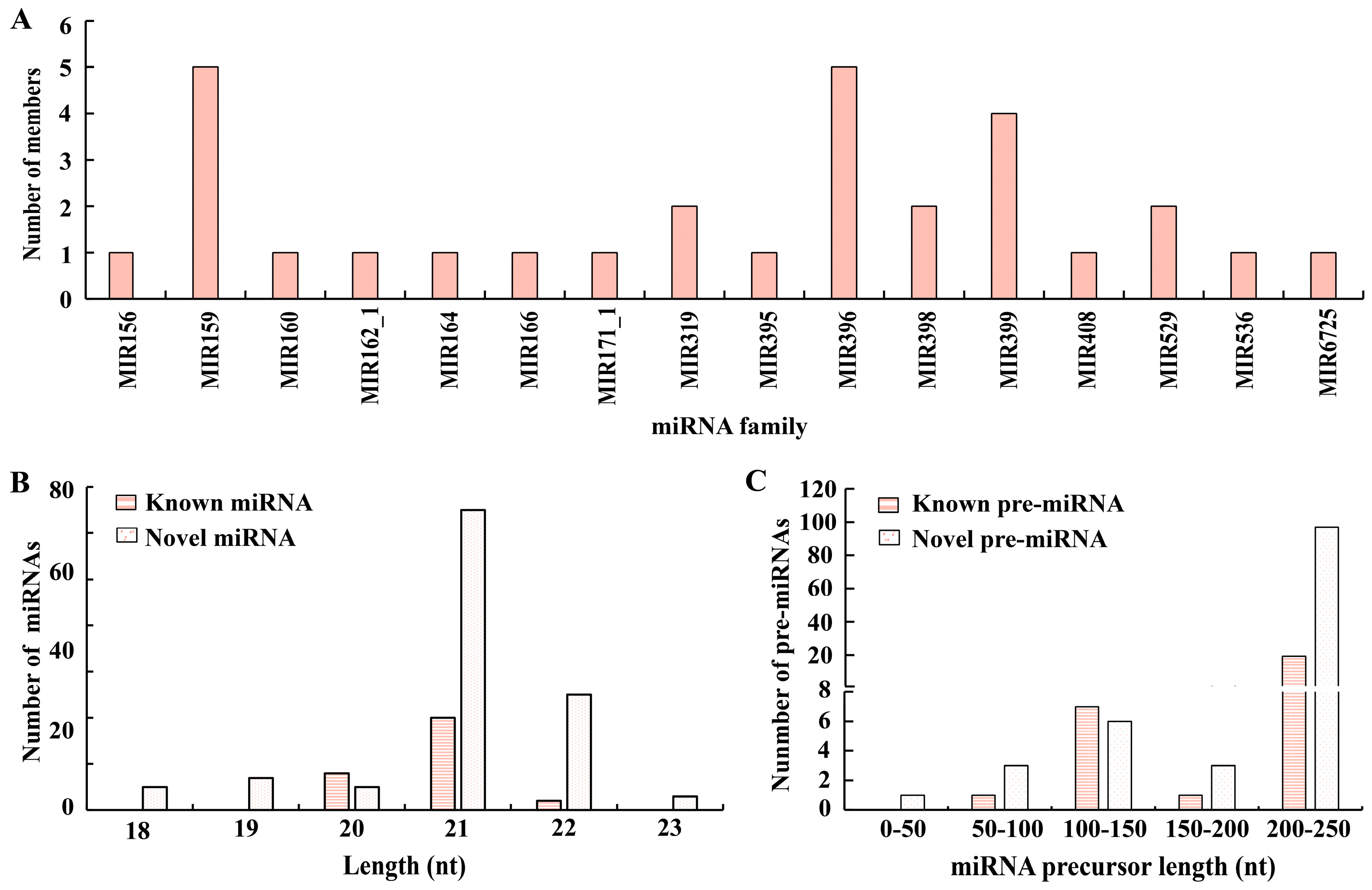
3.2. Classification of the miRNAs
3.3. The Newly Developed miRNAs
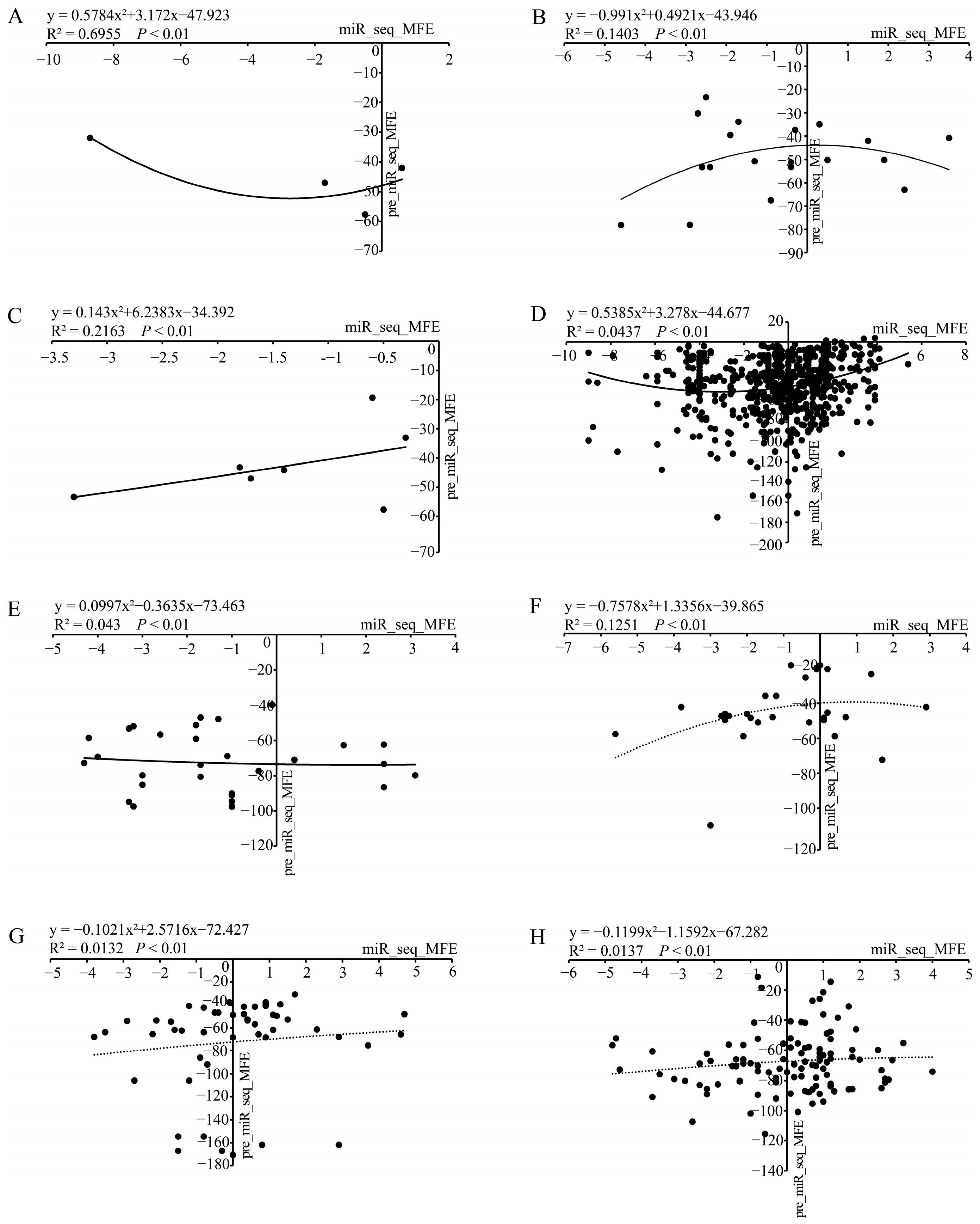
3.4. An Integrated Analysis of the Stability of Chinese Fir miRNAs in Terms of Energy
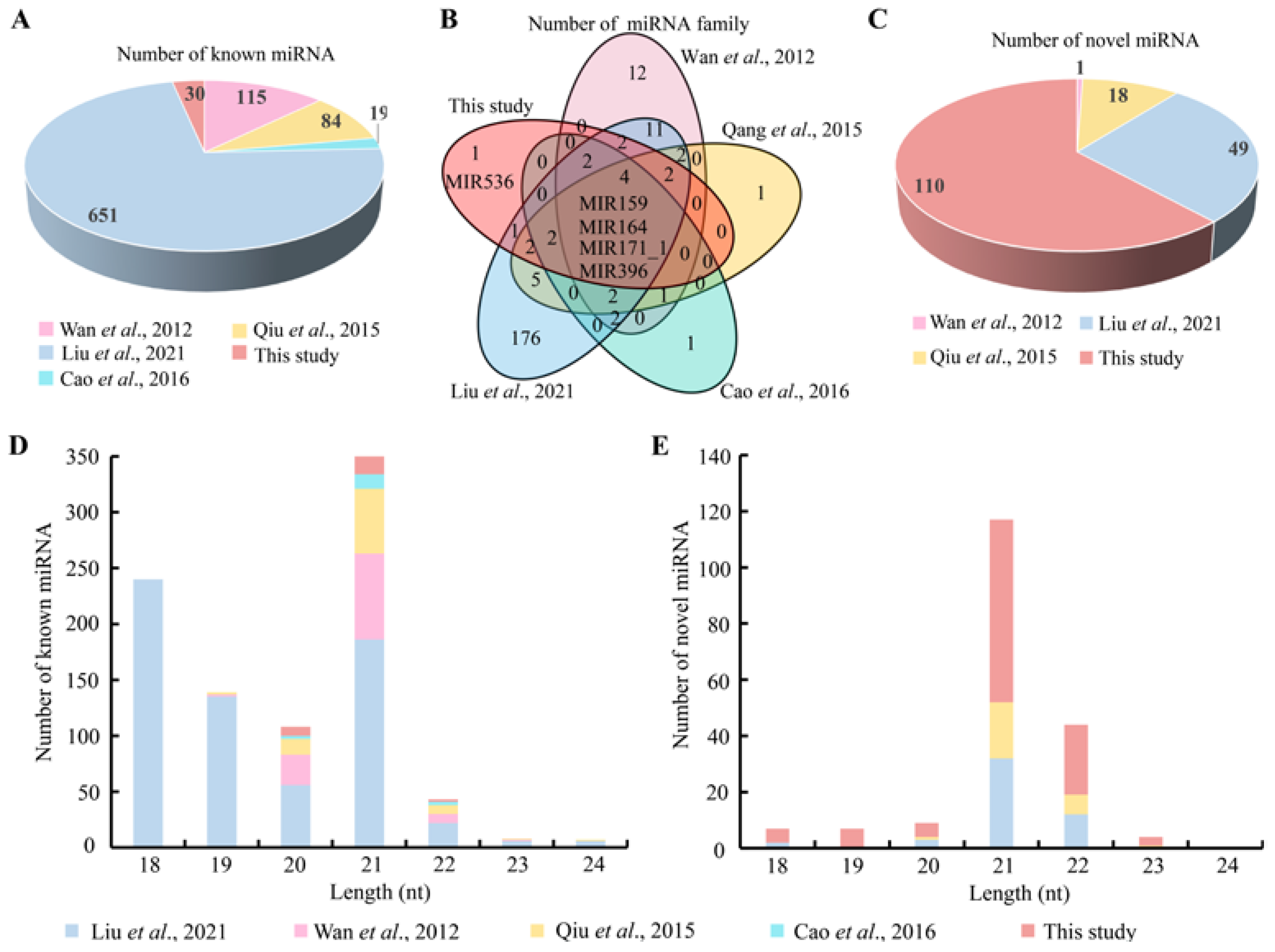
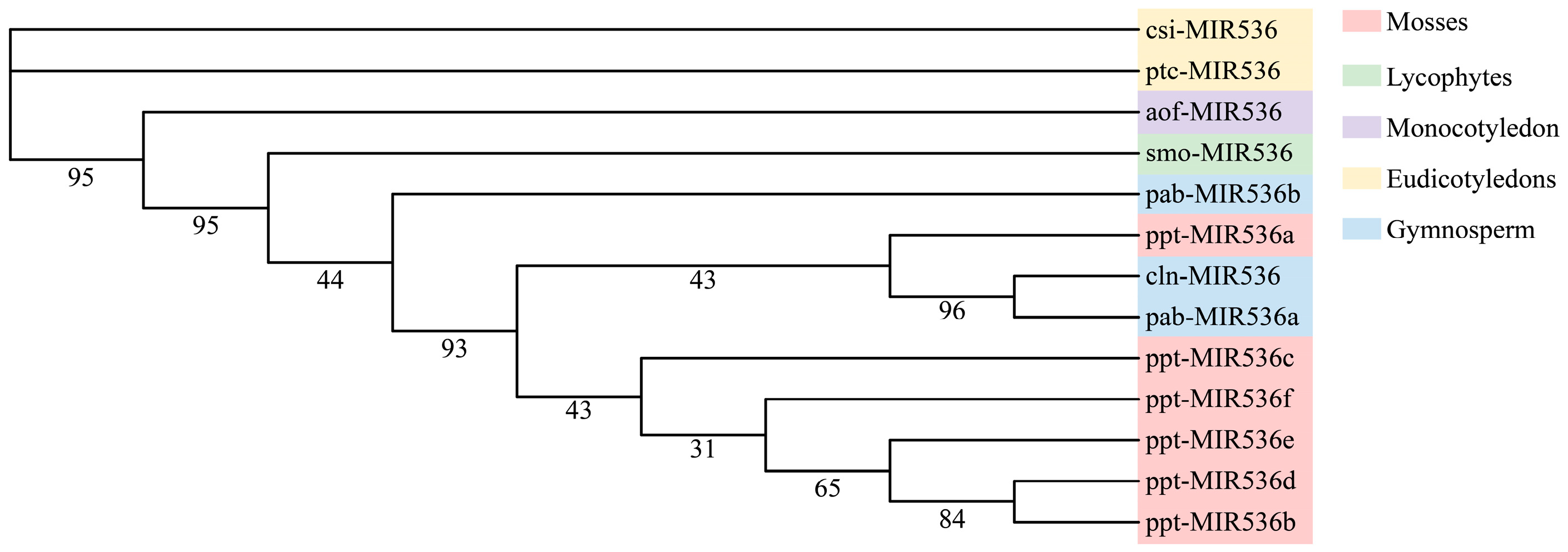
3.5. Phylogenetic Analysis of the Conserved miRNAs between Chinese Fir and Other Plant Species
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J.; et al. Criteria for annotation of plant MicroRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef]
- Bonnet, E.; Wuyts, J.; Rouzé, P.; Van de Peer, Y. Evidence that microRNA precursors, unlike other non-coding RNAs, have lower folding free energies than random sequences. Bioinformatics 2004, 20, 2911–2917. [Google Scholar] [CrossRef]
- Iwakawa, H.O.; Tomari, Y. Molecular insights into microRNA-mediated translational repression in plants. Mol. Cell 2013, 52, 591–601. [Google Scholar] [CrossRef]
- Samad, A.F.A.; Kamaroddin, M.F.; Sajad, M. Cross-kingdom regulation by plant micrornas provides novel insight into gene regulation. Adv. Nutr. 2021, 12, 197–211. [Google Scholar] [CrossRef]
- Yates, L.A.; Norbury, C.J.; Gilbert, R.J.C. The long and short of microRNA. Cell 2013, 153, 516–519. [Google Scholar] [CrossRef]
- Shi, T.; Wang, K.; Yang, P. The evolution of plant microRNAs: Insights from a basal eudicot sacred lotus. Plant J. 2017, 89, 442–457. [Google Scholar] [CrossRef]
- Niu, S.; Li, J.; Bo, W.; Yang, W.; Zuccolo, A.; Giacomello, S.; Chen, X.; Han, F.; Yang, J.; Song, Y.; et al. The Chinese pine genome and methylome unveil key features of conifer evolution. Cell 2022, 185, 204–217. [Google Scholar] [CrossRef]
- Morin, R.D.; Aksay, G.; Dolgosheina, E.; Ebhardt, H.A.; Magrini, V.; Mardis, E.R.; Sahinalp, S.C.; Unrau, P.J. Comparative analysis of the small RNA transcriptomes of Pinus contorta and Oryza sativa. Genome Res. 2008, 18, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Sun, Y.H.; Amerson, H.; Chiang, V.L. MicroRNAs in loblolly pine (Pinus taeda L.) and their association with fusiform rust gall development. Plant J. 2007, 51, 1077–1098. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.; Liu, C.; Yuan, H.W.; Li, P.; Li, Y.; Li, W. Identification and expression profiles of sRNAs and their biogenesis and action-related genes in male and female cones of Pinus tabuliformis. BMC Genom. 2015, 16, 693. [Google Scholar] [CrossRef]
- Shen, T.; Xu, M.; Qi, H.; Feng, Y.; Yang, Z.; Xu, M. Uncovering miRNA-mRNA regulatory modules in developing xylem of Pinus massoniana via small RNA and degradome sequencing. Int. J. Mol. Sci. 2021, 22, 10154. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Xu, J.; Arikit, S.; Meyers, B.C. Extensive families of miRNAs and PHAS loci in Norway Spruce demonstrate the origins of complex phasiRNA networks in seed plants. Mol. Biol. Evol. 2015, 32, 2905–2918. [Google Scholar] [CrossRef] [PubMed]
- Galdino, J.H.; Eguiluz, M.; Guzman, F.; Margis, R. Novel and conserved miRNAs among brazilian pine and other gymnosperms. Front. Genet. 2019, 10, 222. [Google Scholar] [CrossRef]
- Jia, Z.; Zhao, B.; Liu, S.; Lu, Z.; Chang, B.; Jiang, H.; Cui, H.; He, Q.; Li, W.; Jin, B.; et al. Embryo transcriptome and miRNA analyses reveal the regulatory network of seed dormancy in Ginkgo biloba. Tree Physiol. 2021, 41, 571–588. [Google Scholar] [CrossRef]
- Yang, Y.; Hu, X.G.; Zheng, B.; Li, Y.; Wang, T.; Sharma, A.; Yuan, H.; Mao, J.F. Transcriptome-wide identification and characterization of microRNAs and their Targets in a highly adaptable conifer platycladus orientalis. J. Am. Soc. Hortic. Sci. 2022, 147, 7–17. [Google Scholar] [CrossRef]
- Wan, L.C.; Wang, F.; Guo, X.; Lu, S.; Qiu, Z.; Zhao, Y.; Zhang, H.; Lin, J. Identification and characterization of small non-coding RNAs from Chinese fir by high throughput sequencing. BMC Plant Biol. 2012, 12, 146. [Google Scholar] [CrossRef]
- Yang, Y.; Ferguson, D.K.; Liu, B.; Mao, K.; Gao, L.; Zhang, S.; Wan, T.; Rushforth, K.; Zhang, Z. Recent advances on phylogenomics of gymnosperms and a new classification. Plant Divers. 2022, 44, 340–350. [Google Scholar] [CrossRef]
- Shi, J.; Zhen, Y.; Zheng, R.H. Proteome profiling of early seed development in Cunninghamia lanceolata (Lamb.) Hook. J. Exp. Bot. 2010, 61, 2367–2381. [Google Scholar] [CrossRef]
- Zheng, H.; Hu, D.; Wang, R.; Wei, R.; Yan, S. Assessing 62 Chinese Fir (Cunninghamia lanceolata) breeding parents in a 12-year grafted clone test. Forests 2015, 6, 3799–3808. [Google Scholar] [CrossRef]
- Qiu, Z.; Li, X.; Zhao, Y.; Zhang, M.; Wan, Y.; Cao, D.; Lu, S.; Lin, J. Genome-wide analysis reveals dynamic changes in expression of microRNAs during vascular cambium development in Chinese fir, Cunninghamia lanceolata. J. Exp. Bot. 2015, 66, 3041–3054. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Xu, H.; Zhao, Y.; Deng, X.; Liu, Y.; Soppe, W.J.; Lin, J. Transcriptome and degradome sequencing reveals dormancy mechanisms of Cunninghamia lanceolata Seeds. Plant Physiol. 2016, 172, 2347–2362. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Xue, X.; Feng, J.; Cao, D.; Lin, J.; Xu, H. Age-dependent microRNAs in regulation of vascular cambium activity in Chinese fir (Cunninghamia lanceolata). Trees 2021, 35, 1451–1466. [Google Scholar] [CrossRef]
- Charlesworth, D.; Willis, J.H. The genetics of inbreeding depression. Nat. Rev. Genet. 2019, 10, 783–796. [Google Scholar] [CrossRef] [PubMed]
- Rymer, P.D.; Sandiford, M.; Harris, S.A.; Billingham, M.R.; Boshier, D.H. Remnant Pachi-ra quinate pasture trees have greater opportunities to self and suffer reduced reproductive success due to inbreeding depression. Heredity 2015, 115, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Roessler, K.; Muyle, A.; Diez, C.M.; Gaut, G.R.J.; Bousios, A.; Stitzer, M.C.; Seymour, D.K.; Doebley, J.F.; Liu, Q.; Gaut, B.S. The genome-wide dynamics of purging during selfing in maize. Nat. Plants 2019, 5, 980–990. [Google Scholar] [CrossRef]
- Tsuchimatsu, T.; Kakui, H.; Yamazaki, M.; Marona, C.; Tsutsui, H.; Hedhly, A.; Meng, D.; Sato, Y.; Städler, T.; Grossniklaus, U.; et al. Adaptive reduction of male gamete number in the selfing plant Arabidopsis thaliana. Nat. Commun. 2020, 11, 2885. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Wang, J.; Wang, J.; Rausher, M.; Kang, M. Genomic insights into inter- and intra-specific mating system shifts in Primulina. Mol. Ecol. 2022, 31, 5699–5713. [Google Scholar] [CrossRef]
- Wang, R.H.; Hu, D.H.; Zheng, H.Q.; Liu, W.X.; Liang, R.Y.; Yang, B.Q. Genetic Variation Analysis of Flowering Phenology in the 2.5 Generation Seed Orchard of Cunninghamia lanceolata. J. Southwest For. Univ. (Nat. Sci.) 2013, 33, 25–29. [Google Scholar] [CrossRef]
- Wang, Z.M.; Chen, Y.T. An analysis on the combining ability of main growth character in Chinese fir and the application of its heterosis. For. Res. 1988, 1, 614–624. [Google Scholar]
- Chen, Y.T.; He, G.P.; Li, G.X. The effect of inbreeding on seed germination and seedling height growth of Chinese fir. For. Res. 1989, 2, 420–426. [Google Scholar]
- Deng, H.; Hu, D.; Wei, R.; Yan, S.; Wang, R.; Zheng, H. Global transcriptome analysis reveals genes associated with seedling advance growth traits in a selfed family of Chinese fir (Cunninghamia lanceolata). Dendrobiology 2022, 87, 27–46. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008, 36, D154–D158. [Google Scholar] [CrossRef]
- Friedlander, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Fahlgren, N.; Carrington, J.C. miRNA target prediction in plants. Methods Mol. Biol. 2010, 592, 51–57. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.S.; Zeng, H.Q.; Liu, Z.P.; Yang, Z.M. Genome-wide identification of Medicago truncatula microRNAs and their targets reveals their differential regulation by heavy metal. Plant Cell Environ. 2012, 35, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, L.; Yu, M.; Wang, J. Identification and expression analysis of microRNAs during ovule development in rice (Oryza sativa) by deep sequencing. Plant Cell Rep. 2017, 36, 1815–1827. [Google Scholar] [CrossRef]
- Chen, S.Y.; Su, M.H.; Kremling, K.A.; Lepak, N.K.; Romay, M.C.; Sun, Q.; Bradbury, P.J.; Buckler, E.S.; Ku, H.M. Identification of miRNA-eQTLs in maize mature leaf by GWAS. BMC Genom. 2020, 21, 689. [Google Scholar] [CrossRef]
- Dolgosheina, E.V.; Morin, R.D.; Aksay, G.; Sahinalp, S.C.; Magrini, V.; Mardis, E.R.; Mattsson, J.; Unrau, P.J. Conifers have a unique small RNA silencing signature. RNA 2008, 14, 1508–1515. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, J.; Zhu, L.; Xue, J.; Hu, H.; Cui, J.; Xu, J. Identification of microRNAs and their target genes related to needle discoloration of evergreen tree Chinese cedar (Cryptomeria fortunei) in cold winters. Planta 2021, 254, 31. [Google Scholar] [CrossRef] [PubMed]
- Dou, X.; Zhou, Z.; Zhao, L. Identification and expression analysis of miRNAs in germination and seedling growth of Tibetan hulless barley. Genomics 2021, 113, 3735–3749. [Google Scholar] [CrossRef]
- Arro, J.; Cuenca, J.; Yang, Y.; Liang, Z.; Cousins, P.; Zhong, G.Y. A transcriptome analysis of two grapevine populations segregating for tendril phyllotaxy. Hortic. Res. 2017, 4, 17032. [Google Scholar] [CrossRef]
- Hu, H.; Guo, Z.; Yang, J.; Cui, J.; Zhang, Y.; Xu, J. Transcriptome and microRNA sequencing identified miRNAs and target genes in different developmental stages of the vascular cambium in Cryptomeria fortunei Hooibrenk. Front. Plant Sci. 2021, 12, 751–771. [Google Scholar] [CrossRef]
- Yu, Y.; Jia, T.; Chen, X. The ‘how’ and ‘where’ of plant microRNAs. New Phytol. 2017, 216, 1002–1017. [Google Scholar] [CrossRef]
- Lee, Y.; Jeon, K.; Lee, J.T.; Kim, S.; Kim, V.N. MicroRNA maturation: Stepwise processing and subcellular localization. EMBO J. 2002, 21, 4663–4670. [Google Scholar] [CrossRef] [PubMed]
- Barik, S.; SarkarDas, S.; Singh, A.; Gautam, V.; Kumar, P.; Majee, M.; Sarkar, A.K. Phylogenetic analysis reveals conservation and diversification of micro RNA166 genes among diverse plant species. Genomics 2014, 103, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Cha’vez Montes, R.A.; Rosas-Cárdenas, D.F.F.; De Paoli, E.; Accerbi, M.; Rymarquis, L.A.; Mahalingam, G.; Marsch-Martínez, M.; Meyers, B.C.; Green, P.J.; De Folter, S. Sample sequencing of vascular plants demonstrates widespread conservation and divergence of microRNAs. Nat. Commun. 2014, 5, 3722. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Lal, M.; Das, S. Comparative genomics reveals origin of MIR159A-MIR159B paralogy, and complexities of PTGS interaction between miR159 and target GA-MYBs in Brassicaceae. Mol. Genet. Genom. 2019, 294, 693–714. [Google Scholar] [CrossRef]
- Qiu, Z.B.; Yuan, M.M.; Hai, B.Z.; Wang, L.; Zhang, L. Characterization and expression analysis of conserved miRNAs and their targets in Pinus densata. Biol. Plant. 2016, 60, 427–434. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, H.; Huang, R.; Hu, D.; Wang, R.; Wei, R.; Yan, S.; Wu, G.; Sun, Y.; Li, Y.; Zheng, H. Further Mining and Characterization of miRNA Resource in Chinese Fir (Cunninghamia lanceolata). Genes 2022, 13, 2137. https://doi.org/10.3390/genes13112137
Deng H, Huang R, Hu D, Wang R, Wei R, Yan S, Wu G, Sun Y, Li Y, Zheng H. Further Mining and Characterization of miRNA Resource in Chinese Fir (Cunninghamia lanceolata). Genes. 2022; 13(11):2137. https://doi.org/10.3390/genes13112137
Chicago/Turabian StyleDeng, Houyin, Rong Huang, Dehuo Hu, Runhui Wang, Ruping Wei, Su Yan, Guandi Wu, Yuhan Sun, Yun Li, and Huiquan Zheng. 2022. "Further Mining and Characterization of miRNA Resource in Chinese Fir (Cunninghamia lanceolata)" Genes 13, no. 11: 2137. https://doi.org/10.3390/genes13112137
APA StyleDeng, H., Huang, R., Hu, D., Wang, R., Wei, R., Yan, S., Wu, G., Sun, Y., Li, Y., & Zheng, H. (2022). Further Mining and Characterization of miRNA Resource in Chinese Fir (Cunninghamia lanceolata). Genes, 13(11), 2137. https://doi.org/10.3390/genes13112137