
Haemolysis Detection in MicroRNA-Seq from Clinical Plasma Samples

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Information
2.2. Sample Collection
2.3. RNA Extraction and Library Preparation
2.4. Haemolysis Detection by RT-qPCR
2.5. miRNA Annotation and Abundance
2.6. Analysis of Potential Confounding Factors
2.7. Identification of Haemolysis miRNA Signature
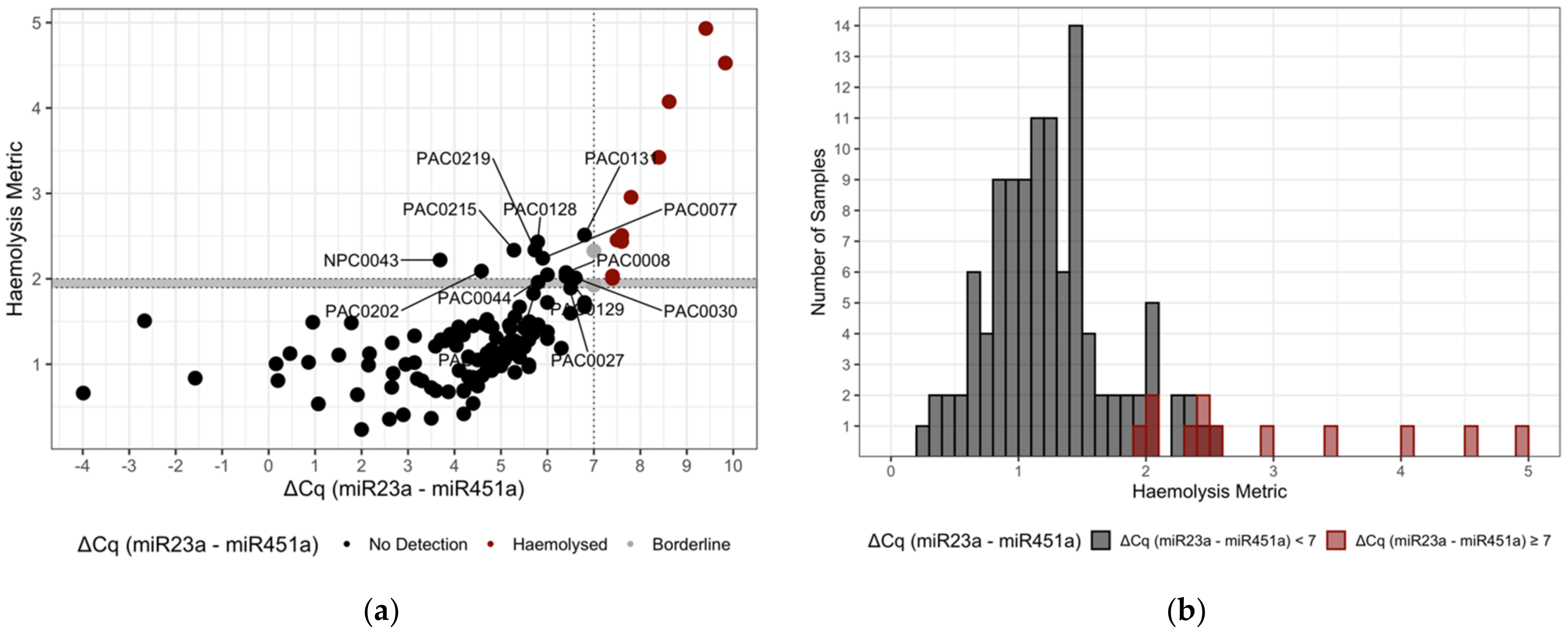
2.8. Classification—Haemolysis Metric
2.9. Data Availability
3. Results
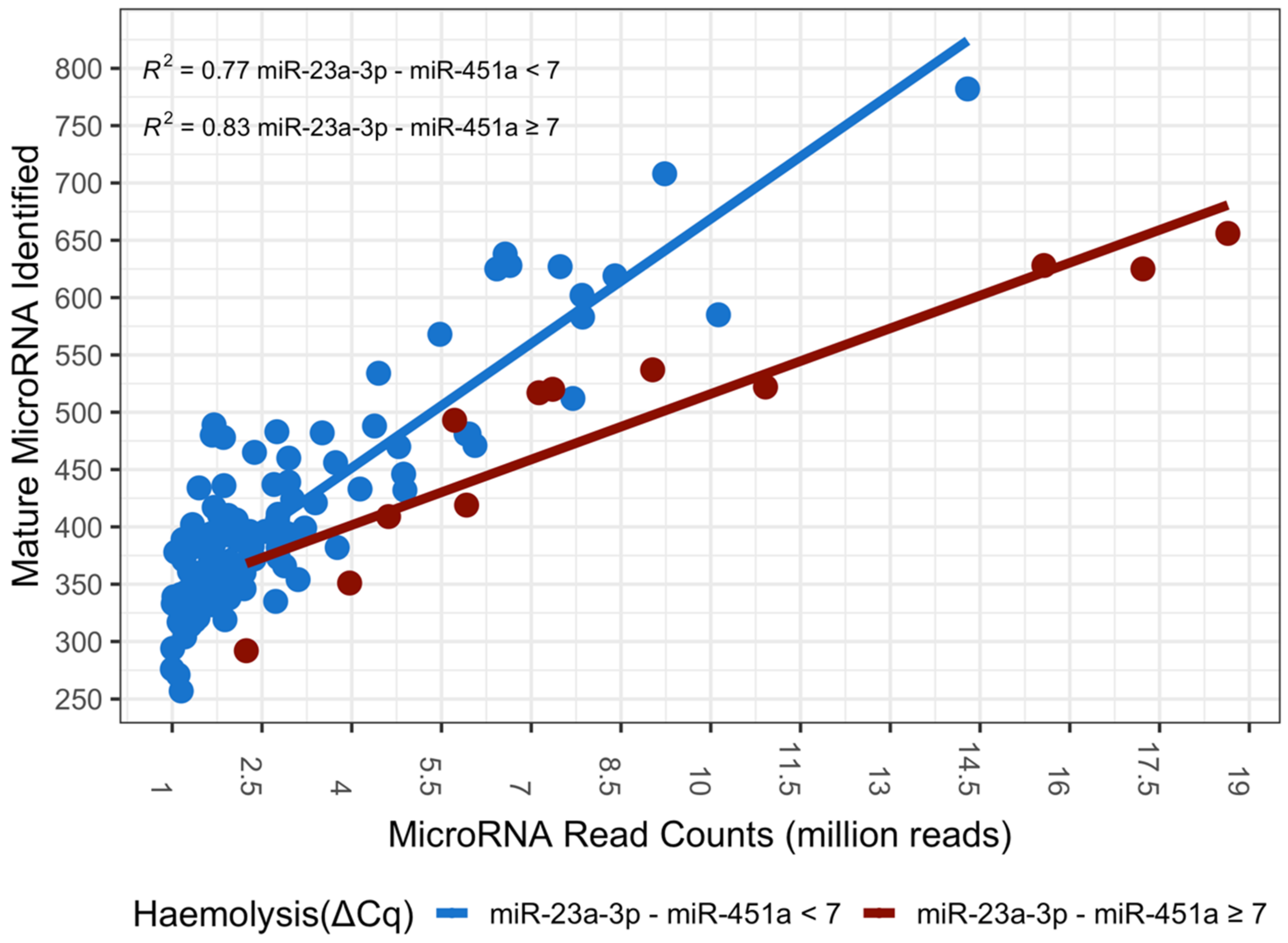
3.1. High-Throughput Sequencing
3.2. MicroRNA Haemolysis Signature Set
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillman, K.A.; Scheer, K.G.; Hackett-Jones, E. Extensive transcriptional responses are co-ordinated by microRNAs as revealed by Exon–Intron Split Analysis (EISA). Nucleic Acids Res. 2019, 47, 8606–8619. Available online: https://academic.oup.com/nar/article-abstract/47/16/8606/5542881 (accessed on 30 March 2022). [CrossRef] [PubMed]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2008, 19, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [Green Version]
- Cortez, M.A.; Calin, G.A. MicroRNA identification in plasma and serum: A new tool to diagnose and monitor diseases. Expert Opin. Biol. Ther. 2009, 9, 703–711. [Google Scholar] [CrossRef]
- Kosaka, N.; Iguchi, H.; Ochiya, T. Circulating microRNA in body fluid: A new potential biomarker for cancer diagnosis and prognosis. Cancer Sci. 2010, 101, 2087–2092. [Google Scholar] [CrossRef]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef] [Green Version]
- Cortez, M.A.; Bueso-Ramos, C.; Ferdin, J.; Lopez-Berestein, G.; Sood, A.K.; Calin, G.A. MicroRNAs in body fluids—The mix of hormones and biomarkers. Nat. Rev. Clin. Oncol. 2011, 8, 467–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.A.; Gandhi, R.; Potla, P.; Keshavarzi, S.; Espin-Garcia, O.; Shestopaloff, K.; Pastrello, C.; Bethune-Waddell, D.; Lively, S.; Perruccio, A.V.; et al. Sequencing identifies a distinct signature of circulating microRNAs in early radiographic knee osteoarthritis. Osteoarthr. Cartil. 2020, 28, 1471–1481. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W. Circulating microRNA biomarker studies: Pitfalls and potential solutions. Clin. Chem. 2015, 61, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.B.; Scelo, G.; Muller, D.C.; Mukeria, A.; Zaridze, D.; Brennan, P. Circulating MicroRNAs as Non-Invasive Biomarkers for Early Detection of Non-Small-Cell Lung Cancer. PLoS ONE 2015, 10, e0125026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschner, M.B.; Kao, S.C.; Edelman, J.J.; Armstrong, N.J.; Vallely, M.P.; van Zandwijk, N.; Reid, G. Haemolysis during sample preparation alters microRNA content of plasma. PLoS ONE 2011, 6, e24145. [Google Scholar] [CrossRef]
- Cheng, H.H.; Yi, H.S.; Kim, Y.; Kroh, E.M.; Chien, J.W.; Eaton, K.D.; Goodman, M.T.; Tait, J.F.; Tewari, M.; Pritchard, C.C. Plasma Processing Conditions Substantially Influence Circulating microRNA Biomarker Levels. PLoS ONE 2013, 8, e64795. [Google Scholar] [CrossRef] [Green Version]
- Kirschner, M.B.; van Zandwijk, N.; Reid, G. Cell-free microRNAs: Potential biomarkers in need of standardized reporting. Front. Genet. 2013, 4, 56. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, C.C.; Kroh, E.; Wood, B.; Arroyo, J.D.; Dougherty, K.J.; Miyaji, M.M.; Tait, J.F.; Tewari, M. Blood cell origin of circulating microRNAs: A cautionary note for cancer biomarker studies. Cancer Prev. Res. 2012, 5, 492–497. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Wong, C.-H.; Song, C.; Heng, K.-S.; Kee, I.H.C.; Tien, S.-L.; Kumarasinghe, P.; Khin, L.-W.; Tan, K.-C. Plasma free hemoglobin: A novel diagnostic test for assessment of the depth of burn injury. Plast. Reconstr. Surg. 2006, 117, 1206–1213. [Google Scholar] [CrossRef] [Green Version]
- Appierto, V.; Callari, M.; Cavadini, E.; Morelli, D.; Daidone, M.G.; Tiberio, P. A lipemia-independent NanoDrop(®)-based score to identify hemolysis in plasma and serum samples. Bioanalysis 2014, 6, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Premachandran, S.; Bagewadikar, R.S.; Poduval, T.B. The use of ELISA to monitor amplified hemolysis by the combined action of osmotic stress and radiation: Potential applications. Radiat. Res. 2005, 163, 351–355. [Google Scholar] [CrossRef]
- Smith, M.D.; Leemaqz, S.Y.; Jankovic-Karasoulos, T.; McCullough, D.; McAninch, D.; Breen, J.; Roberts, C.T.; Pillman, K.A. DraculR: A web based application for in silico haemolysis detection in high throughput small RNA sequencing data. medRxiv 2022. [Google Scholar] [CrossRef]
- Smith, M.D.; Pillman, K.; Jankovic-Karasoulos, T.; McAninch, D.; Wan, Q.; Bogias, K.J.; McCullough, D.; Bianco-Miotto, T.; Breen, J.; Roberts, C.T. Large-scale transcriptome-wide profiling of microRNAs in human placenta and maternal plasma at early to mid gestation. RNA Biol. 2021, 19, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 30 March 2022).
- Didion, J.P.; Martin, M.; Collins, F.S. Atropos: Specific, sensitive, and speedy trimming of sequencing reads. PeerJ 2017, 5, e3720. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Saunders, K.; Bert, A.G.; Dredge, B.K.; Toubia, J.; Gregory, P.A.; Pillman, K.A.; Goodall, G.J.; Bracken, C.P. Insufficiently complex unique-molecular identifiers (UMIs) distort small RNA sequencing. Sci. Rep. 2020, 10, 14593. [Google Scholar] [CrossRef]
- Griffiths-Jones, S. miRBase: microRNA sequences and annotation. Curr. Protoc. Bioinform. 2010, 29, 12.9.1–12.9.10. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. MiRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revelle, W.R. psych: Procedures for Psychological, Psychometric, and Personality Research; Northwestern University: Evanston, IL, USA, 2021. [Google Scholar]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef]
- Giglio, S.; De Nunzio, C.; Cirombella, R.; Stoppacciaro, A.; Faruq, O.; Volinia, S.; Baldassarre, G.; Tubaro, A.; Ishii, H.; Croce, C.M.; et al. A preliminary study of micro-RNAs as minimally invasive biomarkers for the diagnosis of prostate cancer patients. J. Exp. Clin. Cancer Res. 2021, 40, 79. [Google Scholar] [CrossRef]
- Pekacova, A.; Baloun, J.; Sleglova, O.; Růžičková, O.; Vencovský, J.; Pavelka, K.; Šenolt, L. AB0038 Circulating MicroRNAs In Hand Osteoarthritis. Ann. Rheum. Dis. 2021, 80 (Suppl. 1), 1051–1052. [Google Scholar] [CrossRef]
- Sanghavi, M.; Rutherford, J.D. Cardiovascular physiology of pregnancy. Circulation 2014, 130, 1003–1008. [Google Scholar] [CrossRef]
- Shkurnikov, M.Y.; Knyazev, E.N.; Fomicheva, K.A.; Mikhailenko, D.S.; Nyushko, K.M.; Saribekyan, E.K.; Samatov, T.R.; Alekseev, B.Y. Analysis of Plasma microRNA Associated with Hemolysis. Bull. Exp. Biol Med. 2016, 160, 748–750. [Google Scholar] [CrossRef]
- Landoni, E.; Miceli, R.; Callari, M.; Tiberio, P.; Appierto, V.; Angeloni, V.; Mariani, L.; Daidone, M.G. Proposal of supervised data analysis strategy of plasma miRNAs from hybridisation array data with an application to assess hemolysis-related deregulation. BMC Bioinform. 2015, 16, 388. [Google Scholar] [CrossRef] [Green Version]
- McDonald, J.S.; Milosevic, D.; Reddi, H.V.; Grebe, S.K.; Algeciras-Schimnich, A. Analysis of circulating microRNA: Preanalytical and analytical challenges. Clin. Chem. 2011, 57, 833–840. [Google Scholar] [CrossRef] [Green Version]
- Kirschner, M.B.; Edelman, J.J.B.; Kao, S.C.H.; Vallely, M.P.; Van Zandwijk, N.; Reid, G. The impact of hemolysis on cell-free microRNA biomarkers. Front. Genet. 2013, 4, 94. [Google Scholar] [CrossRef] [Green Version]
- Nogales-Gadea, G.; Ramos-Fransi, A.; Suárez-Calvet, X.; Navas, M.; Rojas-García, R.; Mosquera, J.L.; Diaz-Manera, J.; Querol, L.; Gallardo, E.; Illa, I. Analysis of serum miRNA profiles of myasthenia gravis patients. PLoS ONE 2014, 9, e91927. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Fan, G.; Zhang, J.; Wu, C.; Du, Y.; Ye, H.; Li, Z.; Wang, L.; Zhang, Z.; Zhang, L.; et al. A 9-microRNA Signature in Serum Serves as a Noninvasive Biomarker in Early Diagnosis of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 60, 1365–1377. [Google Scholar] [CrossRef] [PubMed]
- Poroyko, V.; Mirzapoiazova, T.; Nam, A.; Mambetsariev, I.; Mambetsariev, B.; Wu, X.; Husain, A.; Vokes, E.E.; Wheeler, D.L.; Salgia, R. Exosomal miRNAs species in the blood of small cell and non-small cell lung cancer patients. Oncotarget 2018, 9, 19793–19806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; He, Y.; Mackowiak, B.; Gao, B. MicroRNAs as regulators, biomarkers and therapeutic targets in liver diseases. Gut 2021, 70, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Li, M.; Huang, Z.; Zhou, X.; Liu, Q.; Xia, T.; Zhu, W. Circulating miR-532-502 cluster derived from chromosome X as biomarkers for diagnosis of breast cancer. Gene 2020, 722, 144104. [Google Scholar] [CrossRef]
- Boeri, M.; Verri, C.; Conte, D.; Roz, L.; Modena, P.; Facchinetti, F.; Calabrò, E.; Croce, C.M.; Pastorino, U.; Sozzi, G. MicroRNA signatures in tissues and plasma predict development and prognosis of computed tomography detected lung cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 3713–3718. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Pregnant | Not Pregnant | |||
|---|---|---|---|---|
| (n = 111) | (n = 10) | |||
| Smoker | Yes | No | Yes | No |
| 48 | 63 | unknown | unknown | |
| Age | 26.9 (21–32) | 24.1 (21–24) | ||
| BMI | 24.3 (21–36) | 23.8 (19–25) | ||
| Gestational age (weeks) | 12.5 (9–16) | Not applicable | ||
| miRNA | Log2FC | Average Expression (log2 CPM) | Adjusted p-Value | Pregnancy Assoc. |
|---|---|---|---|---|
| miR-106b-3p | 1.589 | 8.731 | 8.61 × 10−15 | no |
| miR-140-3p | 1.073 | 10.098 | 2.75 × 10−13 | no |
| miR-142-5p | 0.962 | 10.651 | 4.96 × 10−12 | no |
| miR-532-5p | 1.288 | 7.237 | 4.96 × 10−12 | no |
| miR-17-5p | 0.952 | 7.892 | 7.84 × 10−12 | no |
| miR-19b-3p | 1.128 | 8.696 | 1.93 × 10−09 | no |
| miR-30c-5p | 0.950 | 7.325 | 2.48 × 10−09 | no |
| miR-324-5p | 1.304 | 7.186 | 2.50 × 10−09 | no |
| miR-192-5p | 0.941 | 8.944 | 1.37 × 10−08 | no |
| miR-660-5p | 1.305 | 7.620 | 3.45 × 10−10 | no |
| miR-186-5p | 1.228 | 8.052 | 2.75 × 10−13 | yes |
| miR-425-5p | 1.282 | 11.246 | 4.96 × 10−12 | yes |
| miR-25-3p | 1.212 | 12.939 | 1.26 × 10−11 | yes |
| miR-363-3p | 1.237 | 7.882 | 4.52 × 10−11 | yes |
| miR-183-5p | 1.550 | 9.382 | 9.34 × 10−11 | yes |
| miR-451a | 1.372 | 13.002 | 3.65 × 10−10 | yes |
| miR-182-5p | 1.341 | 10.585 | 2.48 × 10−09 | yes |
| miR-191-5p | 0.929 | 11.790 | 4.68 × 10−09 | yes |
| miR-194-5p | 0.937 | 7.679 | 1.85 × 10−08 | yes |
| miR-20b-5p | 0.932 | 7.430 | 1.96 × 10−08 | yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, M.D.; Leemaqz, S.Y.; Jankovic-Karasoulos, T.; McAninch, D.; McCullough, D.; Breen, J.; Roberts, C.T.; Pillman, K.A. Haemolysis Detection in MicroRNA-Seq from Clinical Plasma Samples. Genes 2022, 13, 1288. https://doi.org/10.3390/genes13071288
Smith MD, Leemaqz SY, Jankovic-Karasoulos T, McAninch D, McCullough D, Breen J, Roberts CT, Pillman KA. Haemolysis Detection in MicroRNA-Seq from Clinical Plasma Samples. Genes. 2022; 13(7):1288. https://doi.org/10.3390/genes13071288
Chicago/Turabian StyleSmith, Melanie D., Shalem Y. Leemaqz, Tanja Jankovic-Karasoulos, Dale McAninch, Dylan McCullough, James Breen, Claire T. Roberts, and Katherine A. Pillman. 2022. "Haemolysis Detection in MicroRNA-Seq from Clinical Plasma Samples" Genes 13, no. 7: 1288. https://doi.org/10.3390/genes13071288
APA StyleSmith, M. D., Leemaqz, S. Y., Jankovic-Karasoulos, T., McAninch, D., McCullough, D., Breen, J., Roberts, C. T., & Pillman, K. A. (2022). Haemolysis Detection in MicroRNA-Seq from Clinical Plasma Samples. Genes, 13(7), 1288. https://doi.org/10.3390/genes13071288