
Bacterial Community Diversity and Bacterial Interaction Network in Eight Mosquito Species



Abstract
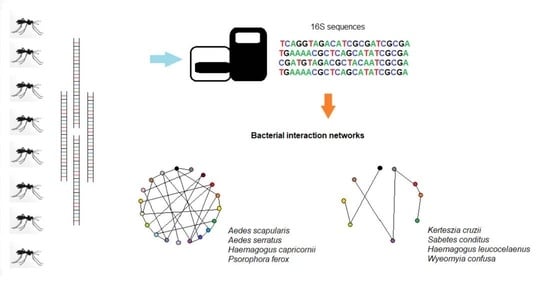
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mosquito Collection and Identification
2.2. Sequencing of 16S rRNA
2.3. Processing of 16S Sequences and Taxonomic Attribution
2.4. Diversity Analyses
2.5. Microbiome Composition Analysis
2.6. PCoA and Heatmap
2.7. Bacterial Interaction Network
3. Results
3.1. 16S Sequences Data
3.2. Bacterial Diversity
3.3. Diversity Analysis
3.4. Microbiome Composition Analysis and Heatmap
3.5. Bacterial Interaction Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harbach, R.E. Culicidae. Mosquito Taxonomic Inventory. 2013. Available online: https://mosquito-taxonomic-inventory.myspecies.info/simpletaxonomy/term/6045 (accessed on 17 July 2022).
- Forattini, O.P. Culicidologia Médica: Identifcaçäo, Biologia e Epidemiologia; Editora da Universidade de São Paulo: São Paulo, Brazil, 2002; Volume 2. [Google Scholar]
- Guedes, M.P. Culicidae (Diptera) no Brasil: Relações entre diversidade, distribuição e enfermidades. Oecol 2012, 16, 283–296. [Google Scholar] [CrossRef]
- Minard, G.; Mavingui, P.; Moro, C.V. Diversity and function of bacterial microbiota in the mosquito holobiont. Parasites Vectors 2013, 6, 146. [Google Scholar] [CrossRef] [PubMed]
- Saab, S.A.; Dohna, H.Z.; Nilsson, L.; Onorati, P.; Nakhleh, J.; Terenius, O.; Osta, M.A. The environment and species affect gut bacteria composition in mosquitoes. Sci. Rep. 2020, 10, 3352. [Google Scholar] [CrossRef] [PubMed]
- Trzebny, A.; Slodkowicz-Kowalska, A.; Björkroth, J.; Dabert, M. Microsporidian Infection in Mosquitoes (Culicidae) Is Associated with Gut Microbiome Composition and Predicted Gut Microbiome Functional Content. Microb. Ecol. 2021, 1–17. [Google Scholar] [CrossRef]
- Coon, K.L.; Brown, M.R.; Strand, M.R. Mosquitoes host communities of bacteria that are essential for development but vary greatly between local habitats. Mol. Ecol. 2016, 25, 5806–5826. [Google Scholar] [CrossRef]
- Cirimotich, C.M.; Ramirez, J.L.; Dimopoulos, G. Native microbiota shape insect vector competence for human pathogens. Cell Host Microbe 2011, 10, 307–310. [Google Scholar] [CrossRef]
- Chouaia, B.; Rossi, P.; Epis, S.; Mosca, M.; Ricci, I.; Damiani, C.; Ulissi, U.; Crotti, E.; Daffonchio, D.; Bandi, C.; et al. Delayed larval development in Anopheles mosquitoes deprived of Asaia bacterial symbionts. BMC Microbiol. 2012, 12, S2. [Google Scholar] [CrossRef]
- Mitraka, E.; Stathopoulos, S.; Siden-Kiamos, I.; Christophides, G.K.; Louis, C. Asaia accelerates development of Anopheles gambiae. Pathog. Glob. Health 2013, 107, 305–311. [Google Scholar] [CrossRef]
- Balaji, S.; Jayachandran, S.; Prabagaran, S.R. Evidence for the natural occurrence of Wolbachia in Aedes aegypti mosquitoes. FEMS Microbiol. Lett. 2019, 366, fnz055. [Google Scholar] [CrossRef]
- Baldini, F.; Segata, N.; Pompon, J.; Marcenac, P.; Shaw, W.R.; Dabiré, R.K.; Diabate, A.; Levashina, E.A.; Catteruccia, F. Evidence of natural Wolbachia infections in field populations of Anopheles gambiae. Nat. Commun. 2014, 5, 3985. [Google Scholar] [CrossRef]
- Wong, M.L.; Liew, J.W.K.; Wong, W.K.; Pramasivan, S.; Hassan, N.M.; Sulaiman, W.Y.W.; Jeyaprakasam, N.K.; Leong, C.S.; Low, V.L.; Vythilingam, I. Natural Wolbachia infection in field-collected Anopheles and other mosquito species from Malaysia. Parasites Vectors 2020, 13, 414. [Google Scholar] [CrossRef] [PubMed]
- Hughes, G.L.; Dodson, B.L.; Johnson, R.M.; Murdock, C.C.; Tsujimoto, H.; Suzuki, Y.; Patt, A.A.; Cui, L.; Nossa, C.W.; Barry, R.M.; et al. Native microbiome impedes vertical transmission of Wolbachia in Anopheles mosquitoes. Proc. Natl. Acad. Sci. USA 2014, 111, 12498–12503. [Google Scholar] [CrossRef]
- Rossi, P.; Ricci, I.; Cappelli, A.; Damiani, C.; Ulissi, U.; Mancini, M.V.; Valzano, M.; Capone, A.; Epis, S.; Crotti, E.; et al. Mutual exclusion of Asaia and Wolbachia in the reproductive organs of mosquito vectors. Parasites Vectors 2015, 8, 278. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Manfredini, F.; Dimopoulos, G. Implication of the mosquito midgut microbiota in the defense against malaria parasites. PLoS Pathog. 2009, 5, e1000423. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.M.; Marinotti, O.; Serrão, D.M.; Correa, L.V.; Katak, R.D.M.; de Oliveira, J.C.; Muniz, V.A.; de Oliveira, M.R.; Neto, J.F.D.N.; Pessoa, M.C.F.; et al. Culturable bacteria associated with Anopheles darlingi and their paratransgenesis potential. Malar. J. 2021, 20, 40. [Google Scholar] [CrossRef]
- Bahia, A.C.; Dong, Y.; Blumberg, B.J.; Mlambo, G.; Tripathi, A.; BenMarzouk-Hidalgo, O.J.; Chandra, R.; Dimopoulos, G. Exploring Anopheles gut bacteria for Plasmodium blocking activity. Environ. Microbiol. 2014, 16, 2980–2994. [Google Scholar] [CrossRef] [PubMed]
- Hegde, S.; Khanipov, K.; Albayrak, L.; Golovko, G.; Pimenova, M.; Saldaña, M.; Rojas, M.M.; Hornett, E.A.; Motl, G.C.; Fredregill, C.; et al. Microbiome Interaction Networks and Community Structure From Laboratory-Reared and Field-Collected Aedes aegypti, Aedes albopictus, and Culex quinquefasciatus Mosquito Vectors. Front. Microbiol. 2018, 9, 2160. [Google Scholar] [CrossRef]
- Tuanudom, R.; Yurayart, N.; Rodkhum, C.; Tiawsirisup, S. Diversity of midgut microbiota in laboratory-colonized and field-collected Aedes albopictus (Diptera: Culicidae): A preliminary study. Heliyon 2021, 7, e08259. [Google Scholar] [CrossRef]
- Kozlova, E.V.; Hegde, S.; Roundy, C.M.; Golovko, G.; Saldaña, M.; Hart, C.E.; Anderson, E.R.; Hornett, E.A.; Khanipov, K.; Popov, V.L.; et al. Microbial interactions in the mosquito gut determine Serratia colonization and blood-feeding propensity. ISME J. 2021, 15, 93–108. [Google Scholar] [CrossRef]
- Bassene, H.; Niang, E.; Fenollar, F.; Dipankar, B.; Doucouré, S.; Ali, E.; Michelle, C.; Raoult, D.; Sokhna, C.; Mediannikov, O.; et al. 16S Metagenomic Comparison of Plasmodium falciparum-Infected and Noninfected Anopheles gambiae and Anopheles funestus Microbiota from Senegal. Am. J. Trop. Med. Hyg. 2018, 99, 1489–1498. [Google Scholar] [CrossRef]
- Caragata, E.P.; Tikhe, C.V.; Dimopoulos, G. Curious entanglements: Interactions between mosquitoes, their microbiota, and arboviruses. Curr. Opin. Virol. 2019, 37, 26–36. [Google Scholar] [CrossRef]
- Scolari, F.; Casiraghi, M.; Bonizzoni, M. Aedes spp. and Their Microbiota: A Review. Front. Microbiol. 2019, 10, 2036. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Cui, C.; Wang, L.; Jacobs-Lorena, M.; Wang, S. Mosquito Microbiota and Implications for Disease Control. Trends Parasitol. 2020, 36, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Zink, S.D.; Van Slyke, G.A.; Palumbo, M.J.; Kramer, L.D.; Ciota, A.T. Exposure to West Nile Virus Increases Bacterial Diversity and Immune Gene Expression in Culex pipiens. Viruses 2015, 7, 5619–5631. [Google Scholar] [CrossRef]
- Balaji, S.; Deepthi, K.; Prabagaran, S.R. Native Wolbachia influence bacterial composition in the major vector mosquito Aedes aegypti. Arch. Microbiol. 2020, 203, 5225–5240. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Raes, J. Microbial interactions: From networks to models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef]
- Faust, K.; Sathirapongsasuti, J.F.; Izard, J.; Segata, N.; Gevers, D.; Raes, J.; Huttenhower, C. Microbial co-occurrence relationships in the human microbiome. PLoS Comput. Biol. 2012, 8, e1002606. [Google Scholar] [CrossRef]
- Hegde, S.; Rasgon, J.L.; Hughes, G.L. The microbiome modulates arbovirus transmission in mosquitoes. Curr. Opin. Virol. 2015, 15, 97–102. [Google Scholar] [CrossRef]
- Sharma, P.; Rani, J.; Chauhan, C.; Kumari, S.; Tevatiya, S.; Das De, T.; Savargaonkar, D.; Pandey, K.C.; Dixit, R. Altered Gut Microbiota and Immunity Defines Plasmodium vivax Survival in Anopheles stephensi. Front. Immunol. 2020, 11, 609. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinform 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Robeson, M.S.; O’Rourke, D.R.; Kaehler, B.D.; Ziemski, M.; Dillon, M.R.; Foster, J.T.; Bokulich, N.A. RESCRIPt: Reproducible sequence taxonomy reference database management for the masses. PLoS Comput. Biol. 2020, 17, e1009581. [Google Scholar] [CrossRef]
- Cardoso, J.; de Almeida, M.A.; dos Santos, E.; da Fonseca, D.F.; Sallum, M.A.; Noll, C.A.; Monteiro, H.A.D.O.; Cruz, A.C.; Carvalho, V.L.; Pinto, E.V.; et al. Yellow fever virus in Haemagogus leucocelaenus and Aedes serratus mosquitoes, southern Brazil, 2008. Emerg. Infect. Dis. 2010, 16, 1918–1924. [Google Scholar] [CrossRef]
- Abreu, F.V.S.; Ribeiro, I.P.; Ferreira-de-Brito, A.; dos Santos, A.A.C.; de Miranda, R.M.; Bonelly, I.D.S.; Neves, M.S.A.S.; Bersot, M.I.; dos Santos, T.P.; Gomes, M.Q.; et al. Haemagogus leucocelaenus and Haemagogus janthinomys are the primary vectors in the major yellow fever outbreak in Brazil, 2016–2018. Emerg. Microbes Infect. 2019, 8, 218–231. [Google Scholar] [CrossRef]
- da Silva, H.; Oliveira, T.M.P.; Sabino, E.C.; Alonso, D.P.; Sallum, M.A.M. Bacterial diversity in Haemagogus leucocelaenus (Diptera: Culicidae) from Vale do Ribeira, São Paulo, Brazil. BMC Microbiol. 2022, 22, 161. [Google Scholar] [CrossRef]
- Curado, I.; Dos Santos Malafronte, R.; de Castro Duarte, A.M.; Kirchgatter, K.; Branquinho, M.S.; Galati, E.A.B. Malaria epidemiology in low-endemicity areas of the Atlantic Forest in the Vale do Ribeira, São Paulo, Brazil. Acta Trop. 2006, 100, 54–62. [Google Scholar] [CrossRef]
- Moreno, E.S.; Barata, R.C.B. Municipalities of higher vulnerability to Sylvatic Yellow Fever occurrence in the São Paulo State, Brazil. Rev. Inst. Med. Trop. São Paulo 2011, 53, 335–339. [Google Scholar] [CrossRef]
- Whitman, L.; Antunes, P.C.A. Studies on the capacity of various Brazilian mosquitoes, representing the Genera Psorophora, Aedes, Mansonia, and Culex, to transmit yellow fever 1. Am. J. Trop. Med. Hyg. 1937, s1-17, 803–823. [Google Scholar] [CrossRef]
- Couto-Lima, D.; Madec, Y.; Bersot, M.I.; Campos, S.S.; Motta, M.D.A.; Santos, F.; Vazeille, M.; Vasconcelos, P.F.D.C.; Lourenço-De-Oliveira, R.; Failloux, A.-B. Potential risk of re-emergence of urban transmission of Yellow Fever virus in Brazil facilitated by competent Aedes populations. Sci. Rep. 2017, 7, 4848. [Google Scholar] [CrossRef] [PubMed]
- Cunha, M.S.; Tubaki, R.M.; de Menezes, R.; Pereira, M.; Caleiro, G.S.; Coelho, E.; Saad, L.D.C.; Fernandes, N.C.C.D.A.; Guerra, J.M.; Nogueira, J.S.; et al. Possible non-sylvatic transmission of yellow fever between non-human primates in São Paulo city, Brazil, 2017–2018. Sci. Rep. 2020, 10, 15751. [Google Scholar] [CrossRef]
- Vasconcelos, P.F.C.; Travassos-da-Rosa, A.P.A.; Pinheiro, F.P.; Shope, R.E.; Degallier, N.; Travassos da Rosa, E.S. Arboviruses pathogenic for man in Brazil. In An Overview of Arbovirology in Brazil and Neighbouring Countries; Travassos-da-Rosa, A.P.A., Vasconcelos, P.F.C., Travassos-da-Rosa, J.F.S., Eds.; Instituto Evandro Chagas: Belém, Brazil, 1998; pp. 72–99. [Google Scholar]
- Duarte, A.M.; Pereira, D.M.; de Paula, M.B.; Fernandes, A.; Urbinatti, P.R.; Ribeiro, A.F.; Mello, M.H.S.; O Matos, M.; Mucci, L.F.; Fernandes, L.N.; et al. Natural infection in anopheline species and its implications for autochthonous malaria in the Atlantic Forest in Brazil. Parasites Vectors 2013, 6, 58. [Google Scholar] [CrossRef] [PubMed]
- Kirchgatter, K.; Tubaki, R.M.; Malafronte, R.; Alves, I.C.; Lima, G.F.M.D.C.; Guimarães, L.D.O.; Zampaulo, R.D.A.; Wunderlich, G. Anopheles (Kerteszia) cruzii (Diptera: Culicidae) in peridomiciliary area during asymptomatic malaria transmission in the Atlantic Forest: Molecular identification of blood-meal sources indicates humans as primary intermediate hosts. Rev. Inst. Med. Trop. São Paulo 2014, 56, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Barrio-Nuevo, K.M.; Cunha, M.S.; Luchs, A.; Fernandes, A.; Rocco, I.M.; Mucci, L.F.; De Souza, R.P.; Medeiros-Sousa, A.R.; Ceretti-Junior, W.; Marrelli, M.T. Detection of Zika and dengue viruses in wild-caught mosquitoes collected during field surveillance in an environmental protection area in São Paulo, Brazil. PLoS ONE 2020, 15, e0227239. [Google Scholar] [CrossRef] [PubMed]
- de Lopes, O.S.; de Sacchetta, L.A.; Francy, D.B.; Jakob, W.L.; Calisher, C.H. Emergence of a new arbovirus disease in Brazil. III. Isolation of Rocio virus from Psorophora Ferox (Humboldt, 1819). Am. J. Epidemiol. 1981, 113, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Mancini, M.V.; Damiani, C.; Accoti, A.; Tallarita, M.; Nunzi, E.; Cappelli, A.; Bozic, J.; Catanzani, R.; Rossi, P.; Valzano, M.; et al. Estimating bacteria diversity in different organs of nine species of mosquito by next generation sequencing. BMC Microbiol. 2018, 18, 126. [Google Scholar] [CrossRef]
- Tchouassi, D.P.; Muturi, E.J.; Arum, S.O.; Kim, C.-H.; Fields, C.J.; Torto, B. Host species and site of collection shape the microbiota of Rift Valley fever vectors in Kenya. PLoS Negl. Trop. Dis. 2019, 13, e0007361. [Google Scholar] [CrossRef]
- Pérez-Ramos, D.W.; Ramos, M.M.; Payne, K.C.; Giordano, B.V.; Caragata, E.P. Collection Time, Location, and Mosquito Species Have Distinct Impacts on the Mosquito Microbiota. Front. Trop. Dis. 2022, 3, 896289. [Google Scholar] [CrossRef]
- Gimonneau, G.; Tchioffo, M.T.; Abate, L.; Boissière, A.; Awono-Ambéné, P.H.; Nsango, S.E.; Christen, R.; Morlais, I. Composition of Anopheles coluzzii and Anopheles gambiae microbiota from larval to adult stages. Infect. Genet. Evol. 2014, 28, 715–724. [Google Scholar] [CrossRef]
- Ibberson, C.B.; Stacy, A.; Fleming, D.; Dees, J.L.; Rumbaugh, K.; Gilmore, M.S.; Whiteley, M. Co-infecting microbes dramatically alter pathogen gene essentiality during polymicrobial infection. Nat. Microbiol. 2017, 2, 17079. [Google Scholar] [CrossRef] [PubMed]
- Gould, A.L.; Zhang, V.; Lamberti, L.; Jones, E.W.; Obadia, B.; Korasidis, N.; Gavryushkin, A.; Carlson, J.M.; Beerenwinkel, N.; Ludington, W.B. Microbiome interactions shape host fitness. Proc. Natl. Acad. Sci. USA 2018, 115, E11951–E11960. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.J.; Hollis, D.G.; Moss, C.W.; English, C.K.; Hall, G.S.; Vincent, J.; Radosevic, J.; A Birkness, K.; Bibb, W.F.; Quinn, F.D. Proposal of Afipia gen. nov., with Afipia felis sp. nov. (formerly the cat scratch disease bacillus), Afipia clevelandensis sp. nov. (formerly the Cleveland Clinic Foundation strain), Afipia broomeae sp. nov., and three unnamed genospecies. J. Clin. Microbiol. 1991, 29, 2450–2460. [Google Scholar] [CrossRef]
- Giladi, M.; Avidor, B.; Kletter, Y.; Abulafia, S.; Slater, L.N.; Welch, D.F.; Brenner, D.J.; Steigerwalt, A.G.; Whitney, A.M.; Ephros, M. Cat scratch disease: The rare role of Afipia felis. J. Clin. Microbiol. 1998, 36, 2499–2502. [Google Scholar] [CrossRef] [PubMed]
- Minard, G.; Tran, F.H.; Raharimalala, F.N.; Hellard, E.; Ravelonandro, P.; Mavingui, P.; Moro, C.V. Prevalence, genomic and metabolic profiles of Acinetobacter and Asaia associated with field-caught Aedes albopictus from Madagascar. FEMS Microbiol. Ecol. 2013, 83, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, T.M.P.; Sanabani, S.S.; Sallum, M.A.M. Bacterial diversity associated with the abdomens of naturally Plasmodium-infected and non-infected Nyssorhynchus darlingi. BMC Microbiol. 2020, 20, 180. [Google Scholar] [CrossRef]
- Muturi, E.J.; Lagos-Kutz, D.; Dunlap, C.; Ramirez, J.L.; Rooney, A.P.; Hartman, G.L.; Fields, C.J.; Rendon, G.; Kim, C.-H. Mosquito microbiota cluster by host sampling location. Parasites Vectors 2018, 11, 468. [Google Scholar] [CrossRef]
- Birnberg, L.; Climent-Sanz, E.; Codoñer, F.M.; Busquets, N. Microbiota Variation Across Life Stages of European Field-Caught Anopheles atroparvus and during Laboratory Colonization: New Insights for Malaria Research. Front. Microbiol. 2021, 12, 3533. [Google Scholar] [CrossRef]
- Capone, A.; Ricci, I.; Damiani, C.; Short, S.M.; Cappelli, A.; Ulissi, U.; Capone, A.; Serrao, A.; Rossi, P.; Amici, A.; et al. Interactions between Asaia, Plasmodium and Anopheles: New insights into mosquito symbiosis and implications in malaria symbiotic control. Parasites Vectors 2013, 6, 182. [Google Scholar] [CrossRef]
- Mancini, M.V.; Damiani, C.; Short, S.M.; Cappelli, A.; Ulissi, U.; Capone, A.; Serrao, A.; Rossi, P.; Amici, A.; Kalogris, C.; et al. Inhibition of Asaia in Adult Mosquitoes Causes Male-Specific Mortality and Diverse Transcriptome Changes. Pathogens 2020, 9, 380. [Google Scholar] [CrossRef]
- Pan, X.; Zhou, G.; Wu, J.; Bian, G.; Lu, P.; Raikhel, A.S.; Xi, Z. Wolbachia induces reactive oxygen species (ROS)-dependent activation of the Toll pathway to control dengue virus in the mosquito Aedes aegypti. Proc. Natl. Acad. Sci. USA 2012, 109, E23–E31. [Google Scholar] [CrossRef] [PubMed]
- Werren, J.H.; Baldo, L.; Clark, M.E. Wolbachia: Master manipulators of invertebrate biology. Nat. Rev. Microbiol. 2008, 6, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Iturbe-Ormaetxe, I.; Walker, T.; O’Neill, S.L. Wolbachia and the biological control of mosquito-borne disease. EMBO Rep. 2011, 12, 508–518. [Google Scholar] [CrossRef]
- Mousson, L.; Zouache, K.; Arias-Goeta, C.; Raquin, V.; Mavingui, P.; Failloux, A.B. The Native Wolbachia Symbionts Limit Transmission of Dengue Virus in Aedes albopictus. PLoS Negl. Trop. Dis. 2012, 6, e1989. [Google Scholar] [CrossRef] [PubMed]
- van den Hurk, A.F.; Hall-Mendelin, S.; Pyke, A.T.; Frentiu, F.D.; McElroy, K.; Day, A.; Higgs, S.; O’Neill, S.L. Impact of Wolbachia on infection with chikungunya and yellow fever viruses in the mosquito vector Aedes aegypti. PLoS Negl. Trop. Dis. 2012, 6, e1892. [Google Scholar] [CrossRef] [PubMed]
- do Nascimento, R.M.; Campolina, T.B.; Chaves, B.A.; Delgado, J.L.F.; Godoy, R.S.M.; Pimenta, P.F.P.; Secundino, N.F.C. The influence of culture-dependent native microbiota in Zika virus infection in Aedes aegypti. Parasites Vectors 2022, 15, 1–14. [Google Scholar] [CrossRef]
- Bai, L.; Wang, L.; Vega-Rodriguez, J.; Wang, G.; Wang, S. A gut symbiotic bacterium Serratia marcescens renders mosquito resistance to Plasmodium infection through activation of mosquito immune responses. Front Microbiol 2019, 10, 1580. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Silva, H.; Oliveira, T.M.P.; Sallum, M.A.M. Bacterial Community Diversity and Bacterial Interaction Network in Eight Mosquito Species. Genes 2022, 13, 2052. https://doi.org/10.3390/genes13112052
da Silva H, Oliveira TMP, Sallum MAM. Bacterial Community Diversity and Bacterial Interaction Network in Eight Mosquito Species. Genes. 2022; 13(11):2052. https://doi.org/10.3390/genes13112052
Chicago/Turabian Styleda Silva, Herculano, Tatiane M. P. Oliveira, and Maria Anice M. Sallum. 2022. "Bacterial Community Diversity and Bacterial Interaction Network in Eight Mosquito Species" Genes 13, no. 11: 2052. https://doi.org/10.3390/genes13112052
APA Styleda Silva, H., Oliveira, T. M. P., & Sallum, M. A. M. (2022). Bacterial Community Diversity and Bacterial Interaction Network in Eight Mosquito Species. Genes, 13(11), 2052. https://doi.org/10.3390/genes13112052