
Identification of a Hypomorphic FANCG Variant in Bernese Mountain Dogs
 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Selection of Cases
2.2. Isolation of DNA
2.3. Library Preparation and Whole Exome Sequencing
2.4. Genotyping Assays
2.5. Measurement of Height at Withers and Weight and Complete Blood Count
2.6. Cell Culture, Plasmids, and Transfection
2.7. Cas9-Mediated Gene Disruption
2.8. Viability Assays
2.9. Immunoblotting
2.10. Statistical Analysis
3. Results
3.1. A Coding Variant in FANCG, Unique to the Genome of BMD is Identified
3.2. The FANCG Variant Is Unique to BMDs
3.3. The FANCG Variant Has a High Allele Frequency in BMDs
3.4. FANCG and MTAP Loci Appear to Segregate Independently in BMDs
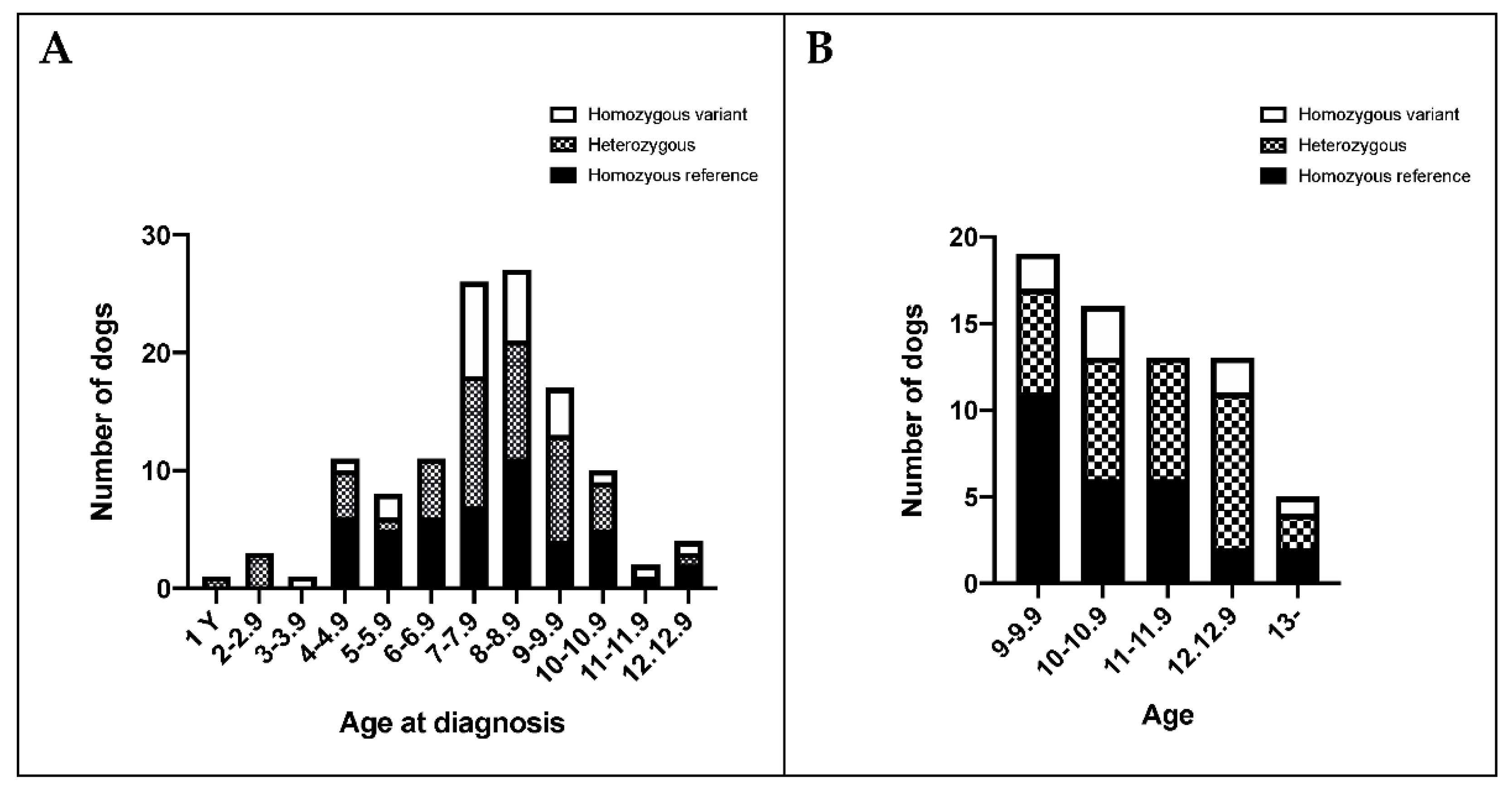
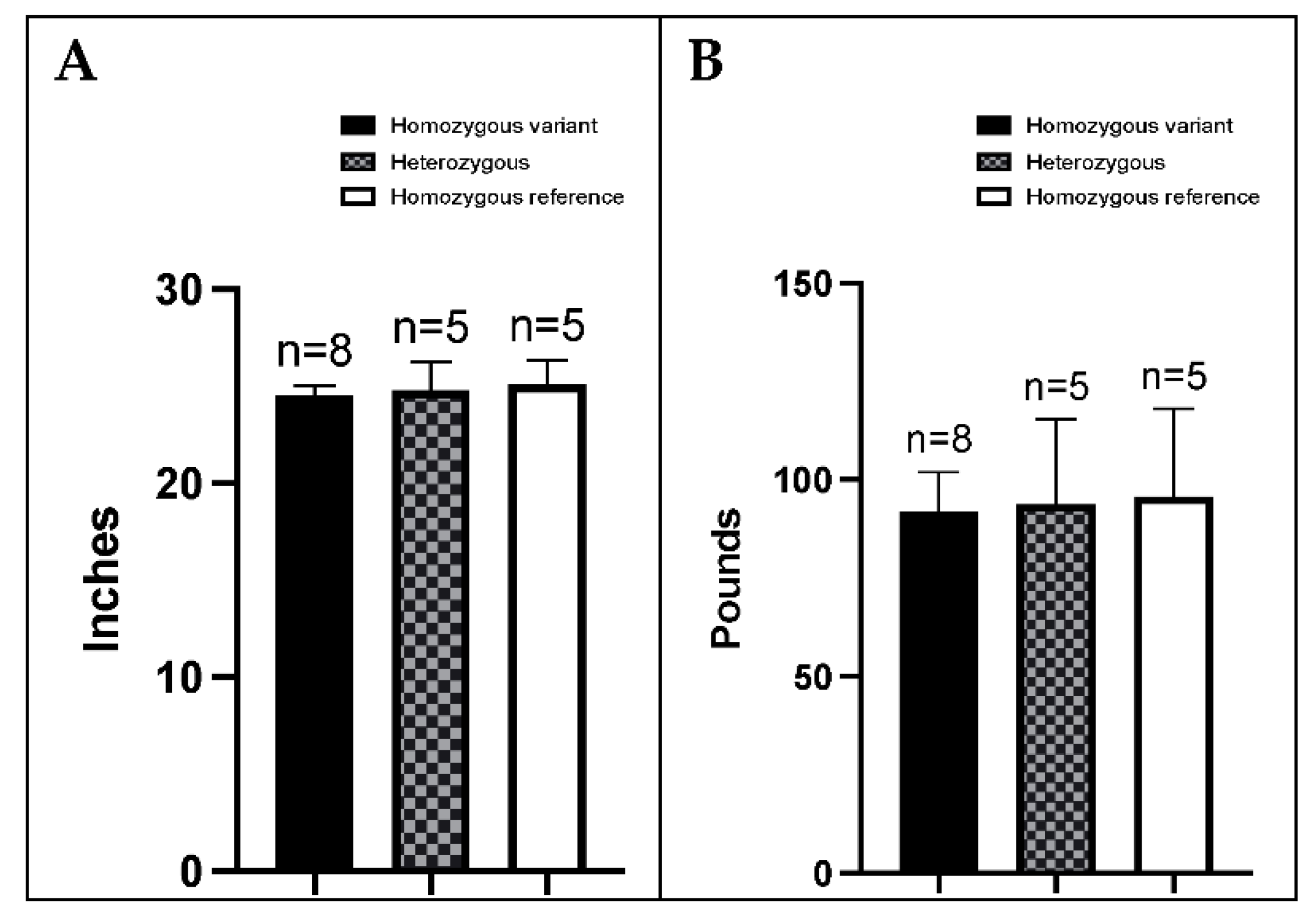
3.5. BMDs Homozygous for the Variant Allele Do Not Display an Overt FA Phenotype
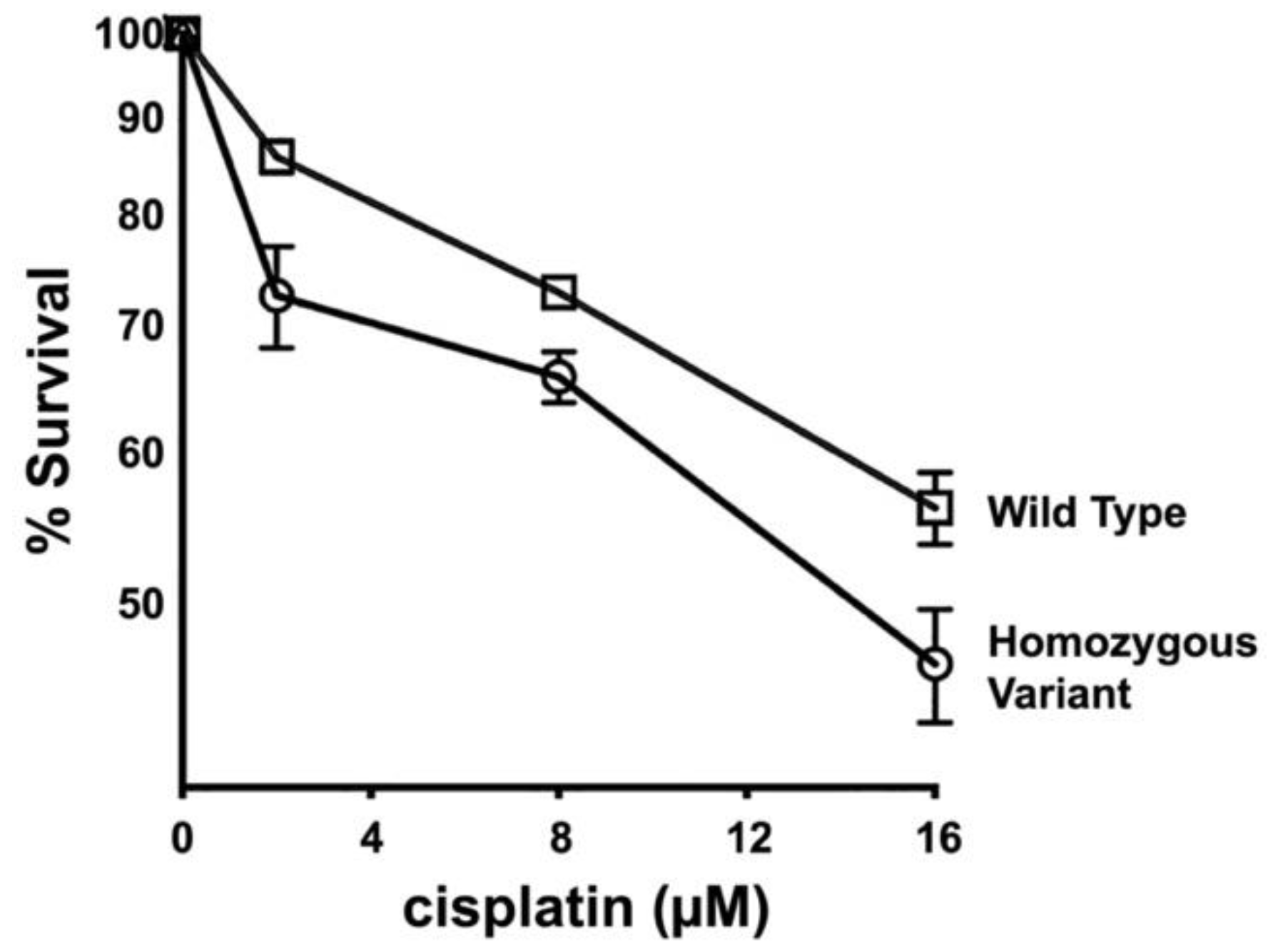
3.6. Primary Fibroblasts from Dogs Homozygous for FANCG Variant Allele Display Increased Cisplatin Sensitivity
3.7. The 465R Substitution Introduced into Human HEK293T Cells Confers Intermediate Cisplatin Sensitivity as Compared to FANCG Deficient 293T Cells
3.8. The 465R Variant Confers Similar Cisplatin Sensitivity as a Human FANCG Hypomorphic Variant That Confers Fanconi’s Anemia in Humans
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schiffman, J.D.; Breen, M. Comparative oncology: What dogs and other species can teach us about humans with cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140231. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.W.; Ostrander, E.A. Domestic dogs and cancer research: A breed-based genomics approach. ILAR J. 2014, 55, 59–68. [Google Scholar] [CrossRef]
- Flisikowski, K.; Flisikowska, T.; Sikorska, A.; Perkowska, A.; Kind, A.; Schnieke, A.; Switonski, M. Germline gene polymorphisms predisposing domestic mammals to carcinogenesis. Vet. Comp. Oncol. 2017, 15, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Abadie, J.; Hedan, B.; Cadieu, E.; De Brito, C.; Devauchelle, P.; Bourgain, C.; Parker, H.G.; Vaysse, A.; Margaritte-Jeannin, P.; Galibert, F.; et al. Epidemiology, pathology, and genetics of histiocytic sarcoma in the Bernese mountain dog breed. J. Hered. 2009, 100 (Suppl. S1), S19–S27. [Google Scholar] [CrossRef] [PubMed]
- Affolter, V.K.; Moore, P.F. Localized and disseminated histiocytic sarcoma of dendritic cell origin in dogs. Vet. Pathol. 2002, 39, 74–83. [Google Scholar] [CrossRef]
- Moore, P.F. A review of histiocytic diseases of dogs and cats. Vet. Pathol. 2014, 51, 167–184. [Google Scholar] [CrossRef]
- Emile, J.F.; Abla, O.; Fraitag, S.; Horne, A.; Haroche, J.; Donadieu, J.; Requena-Caballero, L.; Jordan, M.B.; Abdel-Wahab, O.; Allen, C.E.; et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 2016, 127, 2672–2681. [Google Scholar] [CrossRef]
- Writing Group of the Histiocyte Society. Histiocytosis syndromes in children. Lancet 1987, 1, 208–209. [Google Scholar]
- Shanmugam, V.; Griffin, G.K.; Jacobsen, E.D.; Fletcher, C.D.M.; Sholl, L.M.; Hornick, J.L. Identification of diverse activating mutations of the RAS-MAPK pathway in histiocytic sarcoma. Mod. Pathol. 2019, 32, 830–843. [Google Scholar] [CrossRef]
- Diamond, E.L.; Durham, B.H.; Ulaner, G.A.; Drill, E.; Buthorn, J.; Ki, M.; Bitner, L.; Cho, H.; Young, R.J.; Francis, J.H.; et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature 2019, 567, 521–524. [Google Scholar] [CrossRef]
- Chakraborty, R.; Hampton, O.A.; Shen, X.; Simko, S.J.; Shih, A.; Abhyankar, H.; Lim, K.P.; Covington, K.R.; Trevino, L.; Dewal, N.; et al. Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood 2014, 124, 3007–3015. [Google Scholar] [CrossRef] [PubMed]
- Go, R.S.; Jacobsen, E.; Baiocchi, R.; Buhtoiarov, I.; Butler, E.B.; Campbell, P.K.; Coulter, D.W.; Diamond, E.; Flagg, A.; Goodman, A.M.; et al. Histiocytic Neoplasms, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 1277–1303. [Google Scholar] [CrossRef] [PubMed]
- Goyal, G.; Tazi, A.; Go, R.S.; Rech, K.L.; Picarsic, J.L.; Vassallo, R.; Young, J.R.; Cox, C.W.; Van Laar, J.; Hermiston, M.L.; et al. International expert consensus recommendations for the diagnosis and treatment of Langerhans cell histiocytosis in adults. Blood 2022, 139, 2601–2621. [Google Scholar] [CrossRef] [PubMed]
- Diamond, E.L.; Durham, B.H.; Haroche, J.; Yao, Z.; Ma, J.; Parikh, S.A.; Wang, Z.; Choi, J.; Kim, E.; Cohen-Aubart, F.; et al. Diverse and Targetable Kinase Alterations Drive Histiocytic Neoplasms. Cancer Discov. 2016, 6, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Diamond, E.L.; Subbiah, V.; Lockhart, A.C.; Blay, J.Y.; Puzanov, I.; Chau, I.; Raje, N.S.; Wolf, J.; Erinjeri, J.P.; Torrisi, J.; et al. Vemurafenib for BRAF V600-Mutant Erdheim-Chester Disease and Langerhans Cell Histiocytosis: Analysis of Data From the Histology-Independent, Phase 2, Open-label VE-BASKET Study. JAMA Oncol. 2018, 4, 384–388. [Google Scholar] [CrossRef]
- Thaiwong, T.; Sirivisoot, S.; Takada, M.; Yuzbasiyan-Gurkan, V.; Kiupel, M. Gain-of-function mutation in PTPN11 in histiocytic sarcomas of Bernese Mountain Dogs. Vet. Comp. Oncol. 2018, 16, 220–228. [Google Scholar] [CrossRef]
- Takada, M.; Smyth, L.A.; Thaiwong, T.; Richter, M.; Corner, S.M.; Schall, P.Z.; Kiupel, M.; Yuzbasiyan-Gurkan, V. Activating Mutations in PTPN11 and KRAS in Canine Histiocytic Sarcomas. Genes 2019, 10, 505. [Google Scholar] [CrossRef]
- Hedan, B.; Rault, M.; Abadie, J.; Ulve, R.; Botherel, N.; Devauchelle, P.; Copie-Bergman, C.; Cadieu, E.; Parrens, M.; Alten, J.; et al. PTPN11 mutations in canine and human disseminated histiocytic sarcoma. Int. J. Cancer 2020, 147, 1657–1665. [Google Scholar] [CrossRef]
- Takada, M.; Hix, J.M.L.; Corner, S.; Schall, P.Z.; Kiupel, M.; Yuzbasiyan-Gurkan, V. Targeting MEK in a Translational Model of Histiocytic Sarcoma. Mol. Cancer 2018, 17, 2439–2450. [Google Scholar] [CrossRef]
- Takada, M.; Parys, M.; Gregory-Bryson, E.; Vilar Saavedra, P.; Kiupel, M.; Yuzbasiyan-Gurkan, V. A novel canine histiocytic sarcoma cell line: Initial characterization and utilization for drug screening studies. BMC Cancer 2018, 18, 237. [Google Scholar] [CrossRef]
- Takada, M.; Smyth, L.A.; Hix, J.M.; Corner, S.M.; Kiupel, M.; Yuzbasiyan-Gurkan, V. Development of an Orthotopic Intrasplenic Xenograft Mouse Model of Canine Histiocytic Sarcoma and Its Use in Evaluating the Efficacy of Treatment with Dasatinib. Comp. Med. 2019, 69, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Parmar, K.; D’Andrea, A.; Niedernhofer, L.J. Mouse models of Fanconi anemia. Mutat. Res. 2009, 668, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.L.; D’Andrea, A.D. How the fanconi anemia pathway guards the genome. Annu. Rev. Genet. 2009, 43, 223–249. [Google Scholar] [CrossRef] [PubMed]
- Kee, Y.; D’Andrea, A.D. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010, 24, 1680–1694. [Google Scholar] [CrossRef]
- Garcia-de-Teresa, B.; Rodriguez, A.; Frias, S. Chromosome Instability in Fanconi Anemia: From Breaks to Phenotypic Consequences. Genes 2020, 11, 1528. [Google Scholar] [CrossRef]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef]
- Singh, T.R.; Bakker, S.T.; Agarwal, S.; Jansen, M.; Grassman, E.; Godthelp, B.C.; Ali, A.M.; Du, C.H.; Rooimans, M.A.; Fan, Q.; et al. Impaired FANCD2 monoubiquitination and hypersensitivity to camptothecin uniquely characterize Fanconi anemia complementation group M. Blood 2009, 114, 174–180. [Google Scholar] [CrossRef]
- Meetei, A.R.; de Winter, J.P.; Medhurst, A.L.; Wallisch, M.; Waisfisz, Q.; van de Vrugt, H.J.; Oostra, A.B.; Yan, Z.; Ling, C.; Bishop, C.E.; et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat. Genet. 2003, 35, 165–170. [Google Scholar] [CrossRef]
- Matsushita, N.; Kitao, H.; Ishiai, M.; Nagashima, N.; Hirano, S.; Okawa, K.; Ohta, T.; Yu, D.S.; McHugh, P.J.; Hickson, I.D.; et al. A FancD2-monoubiquitin fusion reveals hidden functions of Fanconi anemia core complex in DNA repair. Mol. Cell 2005, 19, 841–847. [Google Scholar] [CrossRef]
- Hussain, S.; Wilson, J.B.; Medhurst, A.L.; Hejna, J.; Witt, E.; Ananth, S.; Davies, A.; Masson, J.Y.; Moses, R.; West, S.C.; et al. Direct interaction of FANCD2 with BRCA2 in DNA damage response pathways. Hum. Mol. Genet. 2004, 13, 1241–1248. [Google Scholar] [CrossRef]
- Shearin, A.L.; Hedan, B.; Cadieu, E.; Erich, S.A.; Schmidt, E.V.; Faden, D.L.; Cullen, J.; Abadie, J.; Kwon, E.M.; Grone, A.; et al. The MTAP-CDKN2A locus confers susceptibility to a naturally occurring canine cancer. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Hedan, B.; Cadieu, E.; Rimbault, M.; Vaysse, A.; Dufaure de Citres, C.; Devauchelle, P.; Botherel, N.; Abadie, J.; Quignon, P.; Derrien, T.; et al. Identification of common predisposing loci to hematopoietic cancers in four dog breeds. PLoS Genet. 2021, 17, e1009395. [Google Scholar] [CrossRef] [PubMed]
- Ostrander, E.A.; Wayne, R.K. The canine genome. Genome Res. 2005, 15, 1706–1716. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Meek, K.; Jutkowitz, A.; Allen, L.; Glover, J.; Convery, E.; Massa, A.; Mullaney, T.; Stanley, B.; Rosenstein, D.; Bailey, S.M.; et al. SCID dogs: Similar transplant potential but distinct intra-uterine growth defects and premature replicative senescence compared with SCID mice. J. Immunol. 2009, 183, 2529–2536. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef]
- Wang, C.; Lambert, M.W. The Fanconi anemia protein, FANCG, binds to the ERCC1-XPF endonuclease via its tetratricopeptide repeats and the central domain of ERCC1. Biochemistry 2010, 49, 5560–5569. [Google Scholar] [CrossRef]
- Wilson, J.B.; Blom, E.; Cunningham, R.; Xiao, Y.; Kupfer, G.M.; Jones, N.J. Several tetratricopeptide repeat (TPR) motifs of FANCG are required for assembly of the BRCA2/D1-D2-G-X3 complex, FANCD2 monoubiquitylation and phleomycin resistance. Mutat. Res. 2010, 689, 12–20. [Google Scholar] [CrossRef]
- Blom, E.; van de Vrugt, H.J.; de Vries, Y.; de Winter, J.P.; Arwert, F.; Joenje, H. Multiple TPR motifs characterize the Fanconi anemia FANCG protein. DNA Repair 2004, 3, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Fanconi Anemia Mutation Database. Available online: https://www2.rockefeller.edu/fanconi/ (accessed on 10 September 2022).
- Demuth, I.; Wlodarski, M.; Tipping, A.J.; Morgan, N.V.; de Winter, J.P.; Thiel, M.; Grasl, S.; Schindler, D.; D’Andrea, A.D.; Altay, C.; et al. Spectrum of mutations in the Fanconi anaemia group G gene, FANCG/XRCC9. Eur. J. Hum. Genet. 2000, 8, 861–868. [Google Scholar] [CrossRef]
- Jagannathan, V.; Drogemuller, C.; Leeb, T.; Dog Biomedical Variant Database Consortium. A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Anim. Genet. 2019, 50, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Plassais, J.; Kim, J.; Davis, B.W.; Karyadi, D.M.; Hogan, A.N.; Harris, A.C.; Decker, B.; Parker, H.G.; Ostrander, E.A. Whole genome sequencing of canids reveals genomic regions under selection and variants influencing morphology. Nat. Commun. 2019, 10, 1489. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.M.; Thomay, K.; Goehring, G.; Wlodarski, M.; Pastor, V.; Schlegelberger, B.; Schindler, D.; Kratz, C.P.; Niemeyer, C. Mutational Spectrum of Fanconi Anemia Associated Myeloid Neoplasms. Klin. Padiatr. 2017, 229, 329–334. [Google Scholar] [CrossRef]
- Finnie, N.J.; Gottlieb, T.M.; Blunt, T.; Jeggo, P.A.; Jackson, S.P. DNA-dependent protein kinase activity is absent in xrs-6 cells: Implications for site-specific recombination and DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 1995, 92, 320–324. [Google Scholar] [CrossRef]
- Meek, K.; Kienker, L.; Dallas, C.; Wang, W.; Dark, M.J.; Venta, P.J.; Huie, M.L.; Hirschhorn, R.; Bell, T. SCID in Jack Russell terriers: A new animal model of DNA-PKcs deficiency. J. Immunol. 2001, 167, 2142–2150. [Google Scholar] [CrossRef]
- Pace, P.; Mosedale, G.; Hodskinson, M.R.; Rosado, I.V.; Sivasubramaniam, M.; Patel, K.J. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science 2010, 329, 219–223. [Google Scholar] [CrossRef]
- Adamo, A.; Collis, S.J.; Adelman, C.A.; Silva, N.; Horejsi, Z.; Ward, J.D.; Martinez-Perez, E.; Boulton, S.J.; La Volpe, A. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol. Cell 2010, 39, 25–35. [Google Scholar] [CrossRef]
- Chatla, S.; Wilson, A.F.; Pang, Q. Inactivation of the NHEJ Activity of DNA-PKcs Prevents Fanconi Anemia Pre-Leukemic HSC Expansion. Int. J. Stem Cells 2019, 12, 457–462. [Google Scholar] [CrossRef]
- Eccles, L.J.; Bell, A.C.; Powell, S.N. Inhibition of non-homologous end joining in Fanconi Anemia cells results in rescue of survival after interstrand crosslinks but sensitization to replication associated double-strand breaks. DNA Repair 2018, 64, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Thongthip, S.; Conti, B.A.; Lach, F.P.; Smogorzewska, A. Suppression of non-homologous end joining does not rescue DNA repair defects in Fanconi anemia patient cells. Cell Cycle 2020, 19, 2553–2561. [Google Scholar] [CrossRef] [PubMed]
- Neal, J.A.; Dang, V.; Douglas, P.; Wold, M.S.; Lees-Miller, S.P.; Meek, K. Inhibition of homologous recombination by DNA-dependent protein kinase requires kinase activity, is titratable, and is modulated by autophosphorylation. Mol. Cell. Biol. 2011, 31, 1719–1733. [Google Scholar] [CrossRef]
- Fyfe, J.C.; Hemker, S.L.; Venta, P.J.; Stebbing, B.; Giger, U. Selective intestinal cobalamin malabsorption with proteinuria (Imerslund-Grasbeck syndrome) in juvenile Beagles. J. Vet. Intern. Med. 2014, 28, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, J.C.; Hemker, S.L.; Venta, P.J.; Fitzgerald, C.A.; Outerbridge, C.A.; Myers, S.L.; Giger, U. An exon 53 frameshift mutation in CUBN abrogates cubam function and causes Imerslund-Grasbeck syndrome in dogs. Mol. Genet. Metab. 2013, 109, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, J.C.; Madsen, M.; Hojrup, P.; Christensen, E.I.; Tanner, S.M.; de la Chapelle, A.; He, Q.; Moestrup, S.K. The functional cobalamin (vitamin B12)-intrinsic factor receptor is a novel complex of cubilin and amnionless. Blood 2004, 103, 1573–1579. [Google Scholar] [CrossRef]
- Abegglen, L.M.; Caulin, A.F.; Chan, A.; Lee, K.; Robinson, R.; Campbell, M.S.; Kiso, W.K.; Schmitt, D.L.; Waddell, P.J.; Bhaskara, S.; et al. Potential Mechanisms for Cancer Resistance in Elephants and Comparative Cellular Response to DNA Damage in Humans. JAMA 2015, 314, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Oberbeck, N.; Langevin, F.; King, G.; de Wind, N.; Crossan, G.P.; Patel, K.J. Maternal aldehyde elimination during pregnancy preserves the fetal genome. Mol. Cell 2014, 55, 807–817. [Google Scholar] [CrossRef]
- Bakker, S.T.; de Winter, J.P.; te Riele, H. Learning from a paradox: Recent insights into Fanconi anaemia through studying mouse models. Dis. Model Mech. 2013, 6, 40–47. [Google Scholar] [CrossRef]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Daly, M.; Arends, M.J.; Patel, K.J. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 2012, 489, 571–575. [Google Scholar] [CrossRef]
- Langevin, F.; Crossan, G.P.; Rosado, I.V.; Arends, M.J.; Patel, K.J. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 2011, 475, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Dingler, F.A.; Patel, K.J. Genotoxic aldehydes in the hematopoietic system. Blood 2022, 139, 2119–2129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kozono, D.E.; O’Connor, K.W.; Vidal-Cardenas, S.; Rousseau, A.; Hamilton, A.; Moreau, L.; Gaudiano, E.F.; Greenberger, J.; Bagby, G.; et al. TGF-beta Inhibition Rescues Hematopoietic Stem Cell Defects and Bone Marrow Failure in Fanconi Anemia. Cell Stem Cell 2016, 18, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.M.; Mitchell, D.K.; Abdul-Sater, Z.; Chan, K.K.; Sun, Z.; Sheth, A.; He, Y.; Jiang, L.; Yuan, J.; Sharma, R.; et al. Mitotic Errors Promote Genomic Instability and Leukemia in a Novel Mouse Model of Fanconi Anemia. Front. Oncol. 2021, 11, 752933. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Age Range (Years) | FANCG Variant Allele Frequency (%) | FANCG Genotype | ||
|---|---|---|---|---|---|
| Ref/Ref n (%) | Ref/Var n (%) | Var/Var n (%) | |||
| No Cancer (n = 47) | >10 | 39 | 16 (34) | 25 (53) | 6 (13) |
| HS (n = 121) | 1–12.5 | 41 | 47 (39) | 49 (40) | 25 (21) |
| Other Cancers (n = 21) | 3–12 | 33 | 10 (48) | 8 (38) | 3 (14) |
| All Cancers (n = 142) | 1–12.5 | 40 | 57 (40) | 57 (40) | 28 (20) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meek, K.; Yang, Y.-T.; Takada, M.; Parys, M.; Richter, M.; Engleberg, A.I.; Thaiwong, T.; Griffin, R.L.; Schall, P.Z.; Kramer, A.J.; et al. Identification of a Hypomorphic FANCG Variant in Bernese Mountain Dogs. Genes 2022, 13, 1693. https://doi.org/10.3390/genes13101693
Meek K, Yang Y-T, Takada M, Parys M, Richter M, Engleberg AI, Thaiwong T, Griffin RL, Schall PZ, Kramer AJ, et al. Identification of a Hypomorphic FANCG Variant in Bernese Mountain Dogs. Genes. 2022; 13(10):1693. https://doi.org/10.3390/genes13101693
Chicago/Turabian StyleMeek, Katheryn, Ya-Ting Yang, Marilia Takada, Maciej Parys, Marlee Richter, Alexander I. Engleberg, Tuddow Thaiwong, Rachel L. Griffin, Peter Z. Schall, Alana J. Kramer, and et al. 2022. "Identification of a Hypomorphic FANCG Variant in Bernese Mountain Dogs" Genes 13, no. 10: 1693. https://doi.org/10.3390/genes13101693
APA StyleMeek, K., Yang, Y.-T., Takada, M., Parys, M., Richter, M., Engleberg, A. I., Thaiwong, T., Griffin, R. L., Schall, P. Z., Kramer, A. J., & Yuzbasiyan-Gurkan, V. (2022). Identification of a Hypomorphic FANCG Variant in Bernese Mountain Dogs. Genes, 13(10), 1693. https://doi.org/10.3390/genes13101693