
Focused Strategies for Defining the Genetic Architecture of Congenital Heart Defects


Abstract
1. Introduction
2. Phenotypic Considerations
2.1. What Is CHD?
2.2. What Is the Incidence of CHD?
2.3. Tools to Determine CHD Phenotype
3. Genetic Considerations
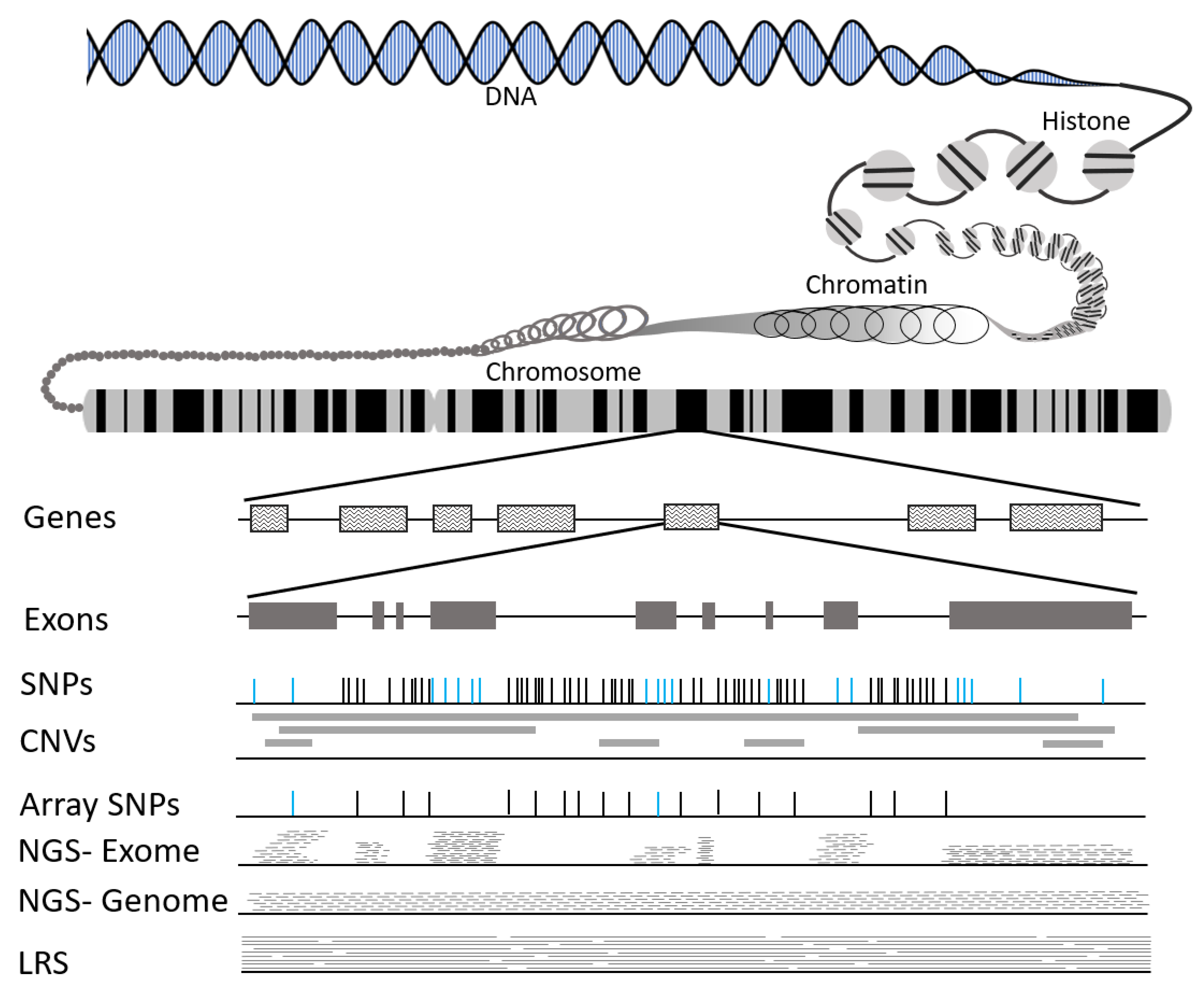
3.1. What Is DNA?
3.2. Types of Genetic Variation
3.3. Tools to Identify Genetic Variation

{kind=link}
| Type of Variant | Description | Consequence | Laboratory Methods | Examples in CHD |
|---|---|---|---|---|
| Single Nucleotide Variation (SNV) | Substitution of single bp for another bp |
| Array +++ NGS +++ LRS +++ | Commonly identified in BAV and HLHS patients, e.g., NOTCH1 [21,22], GATA4 [23], GJA1 [24], and LRP2 [25] |
| Small Insertion/ Deletion (indel) | 1–50 bp duplicated or deleted |
| NGS ++ LRS +++ | SMAD4 [26] ETS1 [27] |
| Tandem Repeats (TR) | Repeats (1–100 bp) occur at single locus |
| LRS +++ | Emerging area of focus. The number of repeats in Fragile X associated with cardiovascular outcomes [33]. |
| Transposable Elements (TE) | Repeats (100 bp–20 kbp) occur at multiple loci |
| LRS +++ | Emerging area of focus. |
| Copy Number Variation (CNV) | Duplication or deletion covering 1 Kb or greater. | Array ++ NGS ++ LRS +++ | Aneuploidies, trisomies, and large SV are often associated with CHD [44,45]. Rare CNV are enriched in both BAV and HLHS [46,47,48,49,50]. |
4. Design and Analytic Considerations
4.1. Strategies to Identify Genetic Variants of Interest
4.2. Analytic Approaches
4.3. Implicating Variants in Disease Etiology
5. Clinical Considerations
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tan, C.M.J.; Lewandowski, A.J. The Transitional Heart: From Early Embryonic and Fetal Development to Neonatal Life. Fetal Diagn. Ther. 2020, 47, 373–386. [Google Scholar] [CrossRef]
- Combs, M.D.; Yutzey, K.E. Heart valve development: Regulatory networks in development and disease. Circ. Res. 2009, 105, 408–421. [Google Scholar] [CrossRef]
- Pierpont, M.E.; Brueckner, M.; Chung, W.K.; Garg, V.; Lacro, R.V.; McGuire, A.L.; Mital, S.; Priest, J.R.; Pu, W.T.; Roberts, A.; et al. American Heart Association Council on Cardiovascular Disease in the Young; Council on Cardiovascular and Stroke Nursing; and Council on Genomic and Precision Medicine. (2018). Genetic basis for congenital heart disease: Revisited: A scientific statement from the American Heart Association. Circulation 2018, 138, e653–e711. [Google Scholar] [CrossRef]
- Hoffman, J.I.; Kaplan, S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002, 39, 1890–1900. [Google Scholar] [CrossRef]
- Cripe, L.; Andelfinger, G.; Martin, L.J.; Shooner, K.; Benson, D.W. Bicuspid aortic valve is heritable. J. Am. Coll. Cardiol. 2004, 44, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.W. The genetics of congenital heart disease: A point in the revolution. Cardiol. Clin. 2002, 20, 385–394. [Google Scholar] [CrossRef]
- Benson, D.W.; Sharkey, A.; Fatkin, D.; Lang, P.; Basson, C.T.; McDonough, B.; Strauss, A.W.; Seidman, J.G.; Seidman, C.E. Reduced penetrance, variable expressivity, and genetic heterogeneity of familial atrial septal defects. Circulation 1998, 97, 2043–2048. [Google Scholar] [CrossRef] [PubMed]
- Devanna, P.; Chen, X.S.; Ho, J.; Gajewski, D.; Smith, S.D.; Gialluisi, A.; Francks, C.; Fisher, S.E.; Newbury, D.F.; Vernes, S.C. Next-gen sequencing identifies non-coding variation disrupting miRNA-binding sites in neurological disorders. Mol. Psychiatry 2018, 23, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Lupski, J.R. Non-coding genetic variants in human disease. Hum. Mol. Genet. 2015, 24, R102–R110. [Google Scholar] [CrossRef] [PubMed]
- Moriya, T.; Yamamura, M.; Kiga, D. Effects of downstream genes on synthetic genetic circuits. BMC Syst. Biol. 2014, 8, 1–10. [Google Scholar] [CrossRef]
- Smemo, S.; Tena, J.J.; Kim, K.H.; Gamazon, E.R.; Sakabe, N.J.; Gomez-Marin, C.; Aneas, I.; Credidio, F.L.; Sobreira, D.R.; Wasserman, N.F.; et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 2014, 507, 371–375. [Google Scholar] [CrossRef]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Richter, F.; Morton, S.U.; Kim, S.W.; Kitaygorodsky, A.; Wasson, L.K.; Chen, K.M.; Zhou, J.; Qi, H.; Patel, N.; DePalma, S.R.; et al. Genomic analyses implicate noncoding de novo variants in congenital heart disease. Nat. Genet. 2020, 52, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Ferguson-Smith, M.A. History and evolution of cytogenetics. Mol. Cytogenet. 2015, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bumgarner, R. Overview of DNA microarrays: Types, applications, and their future. Curr. Protoc. Mol. Biol. 2013, 101, 22.1.1–22.1.11. [Google Scholar] [CrossRef]
- A haplotype map of the human genome. Nature 2005, 437, 1299–1320. [CrossRef]
- Carter, N.P. Methods and strategies for analyzing copy number variation using DNA microarrays. Nat. Genet. 2007, 39, S16–S21. [Google Scholar] [CrossRef]
- Coughlin, C.R.; Scharer, G.H.; Shaikh, T.H. Clinical impact of copy number variation analysis using high-resolution microarray technologies: Advantages, limitations and concerns. Genome Med. 2012, 4, 1–12. [Google Scholar] [CrossRef]
- Shen, H.; Li, J.; Zhang, J.; Xu, C.; Jiang, Y.; Wu, Z.; Zhao, F.; Liao, L.; Chen, J.; Lin, Y.; et al. Comprehensive characterization of human genome variation by high coverage whole-genome sequencing of forty four Caucasians. PLoS ONE 2013, 8, e59494. [Google Scholar] [CrossRef] [PubMed]
- Vidal, E.A.; Moyano, T.C.; Bustos, B.I.; Perez-Palma, E.; Moraga, C.; Riveras, E.; Montecinos, A.; Azocar, L.; Soto, D.C.; Vidal, M.; et al. Whole Genome Sequence, Variant Discovery and Annotation in Mapuche-Huilliche Native South Americans. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Garg, V.; Muth, A.N.; Ransom, J.F.; Schluterman, M.K.; Barnes, R.; King, I.N.; Grossfeld, P.D.; Srivastava, D. Mutations in NOTCH1 cause aortic valve disease. Nature 2005, 437, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Foffa, I.; Ali, L.A.; Panesi, P.; Mariani, M.; Festa, P.; Botto, N.; Vecoli, C.; Andreassi, M.G. Sequencing of NOTCH1, GATA5, TGFBR1 and TGFBR2 genes in familial cases of bicuspid aortic valve. BMC Med. Genet. 2013, 14, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhou, W.; Jiao, J.; Nielsen, J.B.; Mathis, M.R.; Heydarpour, M.; Lettre, G.; Folkersen, L.; Prakash, S.; Schurmann, C.; et al. Protein-altering and regulatory genetic variants near GATA4 implicated in bicuspid aortic valve. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Dasgupta, C.; Martinez, A.M.; Zuppan, C.W.; Shah, M.M.; Bailey, L.L.; Fletcher, W.H. Identification of connexin43 (alpha1) gap junction gene mutations in patients with hypoplastic left heart syndrome by denaturing gradient gel electrophoresis (DGGE). Mutat. Res. 2001, 479, 173–186. [Google Scholar] [CrossRef]
- Theis, J.L.; Vogler, G.; Missinato, M.A.; Li, X.; Nielsen, T.; Zeng, X.I.; Martinez-Fernandez, A.; Walls, S.M.; Kervadec, A.; Kezos, J.N.; et al. Patient-specific genomics and cross-species functional analysis implicate LRP2 in hypoplastic left heart syndrome. eLife 2020, 9, e59554. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Park, J.S.; Jang, S.Y.; Park, S.H.; Kim, J.W.; Ki, C.S.; Kim, D.K. A novel SMAD6 variant in a patient with severely calcified bicuspid aortic valve and thoracic aortic aneurysm. Mol. Genet. Genom. Med. 2019, 7, e620. [Google Scholar] [CrossRef] [PubMed]
- Tootleman, E.; Malamut, B.; Akshoomoff, N.; Mattson, S.N.; Hoffman, H.M.; Jones, M.C.; Printz, B.; Shiryaev, S.A.; Grossfeld, P. Partial Jacobsen syndrome phenotype in a patient with a de novo frameshift mutation in the ETS1 transcription factor. Mol. Case Stud. 2019, 5, a004010. [Google Scholar] [CrossRef]
- Torresen, O.K.; Star, B.; Mier, P.; Andrade-Navarro, M.A.; Bateman, A.; Jarnot, P.; Gruca, A.; Grynberg, M.; Kajava, A.V.; Promponas, V.J.; et al. Tandem repeats lead to sequence assembly errors and impose multi-level challenges for genome and protein databases. Nucleic Acids Res. 2019, 47, 10994–11006. [Google Scholar] [CrossRef]
- Usdin, K. The biological effects of simple tandem repeats: Lessons from the repeat expansion diseases. Genome Res. 2008, 18, 1011–1019. [Google Scholar] [CrossRef]
- Tang, H.; Kirkness, E.F.; Lippert, C.; Biggs, W.H.; Fabani, M.; Guzman, E.; Ramakrishnan, S.; Lavrenko, V.; Kakaradov, B.; Hou, C.; et al. Profiling of Short-Tandem-Repeat Disease Alleles in 12,632 Human Whole Genomes. Am. J. Hum. Genet. 2017, 101, 700–715. [Google Scholar] [CrossRef]
- Sun, J.H.; Zhou, L.; Emerson, D.J.; Phyo, S.A.; Titus, K.R.; Gong, W.; Gilgenast, T.G.; Beagan, J.A.; Davidson, B.L.; Tassone, F.; et al. Disease-Associated Short Tandem Repeats Co-localize with Chromatin Domain Boundaries. Cell 2018, 175, 224–238. [Google Scholar] [CrossRef] [PubMed]
- Sulovari, A.; Li, R.; Audano, P.A.; Porubsky, D.; Vollger, M.R.; Logsdon, G.A.; Variation, C.H.G.S.; Warren, W.C.; Pollen, A.A.; Chaisson, M.J.P.; et al. Human-specific tandem repeat expansion and differential gene expression during primate evolution. Proc. Natl. Acad. Sci. USA 2019, 116, 23243–23253. [Google Scholar] [CrossRef] [PubMed]
- Tassanakijpanich, N.; Cohen, J.; Cohen, R.; Srivatsa, U.N.; Hagerman, R.J. Cardiovascular Problems in the Fragile X Premutation. Front. Genet. 2020, 11, 1244. [Google Scholar] [CrossRef]
- Kazazian, H.H., Jr.; Wong, C.; Youssoufian, H.; Scott, A.F.; Phillips, D.G.; Antonarakis, S.E. A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature 1988, 332, 164–166. [Google Scholar] [CrossRef]
- Ye, M.; Goudot, C.; Hoyler, T.; Lemoine, B.; Amigorena, S.; Zueva, E. Specific subfamilies of transposable elements contribute to different domains of T lymphocyte enhancers. Proc. Natl. Acad. Sci. USA 2020, 117, 7905–7916. [Google Scholar] [CrossRef]
- Faulkner, G.J.; Billon, V. L1 retrotransposition in the soma: A field jumping ahead. Mob. DNA 2018, 9, 1–18. [Google Scholar] [CrossRef]
- Pehrsson, E.C.; Choudhary, M.N.K.; Sundaram, V.; Wang, T. The epigenomic landscape of transposable elements across normal human development and anatomy. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Diehl, A.G.; Ouyang, N.; Boyle, A.P. Transposable elements contribute to cell and species-specific chromatin looping and gene regulation in mammalian genomes. Nat. Commun. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Abel, H.J.; Larson, D.E.; Regier, A.A.; Chiang, C.; Das, I.; Kanchi, K.L.; Layer, R.M.; Neale, B.M.; Salerno, W.J.; Reeves, C.; et al. Mapping and characterization of structural variation in 17,795 human genomes. Nature 2020, 583, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.; Scott, A.J.; Davis, J.R.; Tsang, E.K.; Li, X.; Kim, Y.; Hadzic, T.; Damani, F.N.; Ganel, L.; Consortium, G.T.; et al. The impact of structural variation on human gene expression. Nat. Genet. 2017, 49, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Zhao, X.; Benton, M.L.; Perumal, T.; Collins, R.L.; Hoffman, G.E.; Johnson, J.S.; Sloofman, L.; Wang, H.Z.; Stone, M.R.; et al. Functional annotation of rare structural variation in the human brain. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Alonge, M.; Wang, X.; Benoit, M.; Soyk, S.; Pereira, L.; Zhang, L.; Suresh, H.; Ramakrishnan, S.; Maumus, F.; Ciren, D.; et al. Major Impacts of Widespread Structural Variation on Gene Expression and Crop Improvement in Tomato. Cell 2020, 182, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Costain, G.; Silversides, C.K.; Bassett, A.S. The importance of copy number variation in congenital heart disease. NPJ Genom. Med. 2016, 1, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chang, X.; Glessner, J.; Qu, H.; Tian, L.; Li, D.; Nguyen, K.; Sleiman, P.M.A.; Hakonarson, H. Association of Rare Recurrent Copy Number Variants with Congenital Heart Defects Based on Next-Generation Sequencing Data from Family Trios. Front. Genet. 2019, 10, 819. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.K.; Bondy, C.A.; Maslen, C.L.; Silberbach, M.; Lin, A.E.; Perrone, L.; Limongelli, G.; Michelena, H.I.; Bossone, E.; Citro, R.; et al. Autosomal and X chromosome structural variants are associated with congenital heart defects in Turner syndrome: The NHLBI GenTAC registry. Am. J. Med. Genet. A 2016, 170, 3157–3164. [Google Scholar] [CrossRef]
- Hitz, M.P.; Lemieux-Perreault, L.P.; Marshall, C.; Feroz-Zada, Y.; Davies, R.; Yang, S.W.; Lionel, A.C.; D’Amours, G.; Lemyre, E.; Cullum, R.; et al. Rare copy number variants contribute to congenital left-sided heart disease. PLoS Genet. 2012, 8, e1002903. [Google Scholar] [CrossRef]
- Glidewell, S.C.; Miyamoto, S.D.; Grossfeld, P.D.; Clouthier, D.E.; Coldren, C.D.; Stearman, R.S.; Geraci, M.W. Transcriptional Impact of Rare and Private Copy Number Variants in Hypoplastic Left Heart Syndrome. Clin. Transl. Sci. 2015, 8, 682–689. [Google Scholar] [CrossRef]
- Carey, A.S.; Liang, L.; Edwards, J.; Brandt, T.; Mei, H.; Sharp, A.J.; Hsu, D.T.; Newburger, J.W.; Ohye, R.G.; Chung, W.K.; et al. Effect of copy number variants on outcomes for infants with single ventricle heart defects. Circ. Cardiovasc. Genet. 2013, 6, 444–451. [Google Scholar] [CrossRef]
- Luyckx, I.; Kumar, A.A.; Reyniers, E.; Dekeyser, E.; Vanderstraeten, K.; Vandeweyer, G.; Wunnemann, F.; Preuss, C.; Mazzella, J.M.; Goudot, G.; et al. Copy number variation analysis in bicuspid aortic valve-related aortopathy identifies TBX20 as a contributing gene. Eur. J. Hum. Genet. 2019, 27, 1033–1043. [Google Scholar] [CrossRef]
- Prakash, S.; Kuang, S.Q.; GenTAC Registry Investigators; Regalado, E.; Guo, D.; Milewicz, D. Recurrent Rare Genomic Copy Number Variants and Bicuspid Aortic Valve Are Enriched in Early Onset Thoracic Aortic Aneurysms and Dissections. PLoS ONE 2016, 11, e0153543. [Google Scholar] [CrossRef]
- Behjati, S.; Tarpey, P.S. What is next generation sequencing? Arch. Dis. Child. Educ. Pract. 2013, 98, 236–238. [Google Scholar] [CrossRef] [PubMed]
- Kanzi, A.M.; San, J.E.; Chimukangara, B.; Wilkinson, E.; Fish, M.; Ramsuran, V.; de Oliveira, T. Next Generation Sequencing and Bioinformatics Analysis of Family Genetic Inheritance. Front. Genet. 2020, 11, 1250. [Google Scholar] [CrossRef] [PubMed]
- Rehm, H.L.; Bale, S.J.; Bayrak-Toydemir, P.; Berg, J.S.; Brown, K.K.; Deignan, J.L.; Friez, M.J.; Funke, B.H.; Hegde, M.R.; Lyon, E. ACMG clinical laboratory standards for next-generation sequencing. Genet. Med. 2013, 15, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Trudso, L.C.; Andersen, J.D.; Jacobsen, S.B.; Christiansen, S.L.; Congost-Teixidor, C.; Kampmann, M.L.; Morling, N. A comparative study of single nucleotide variant detection performance using three massively parallel sequencing methods. PLoS ONE 2020, 15, e0239850. [Google Scholar] [CrossRef]
- Belkadi, A.; Bolze, A.; Itan, Y.; Cobat, A.; Vincent, Q.B.; Antipenko, A.; Shang, L.; Boisson, B.; Casanova, J.L.; Abel, L. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. USA 2015, 112, 5473–5478. [Google Scholar] [CrossRef]
- Merker, J.D.; Wenger, A.M.; Sneddon, T.; Grove, M.; Zappala, Z.; Fresard, L.; Waggott, D.; Utiramerur, S.; Hou, Y.; Smith, K.S.; et al. Long-read genome sequencing identifies causal structural variation in a Mendelian disease. Genet. Med. 2018, 20, 159–163. [Google Scholar] [CrossRef]
- Mantere, T.; Kersten, S.; Hoischen, A. Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 2019, 10, 426. [Google Scholar] [CrossRef] [PubMed]
- Ballouz, S.; Dobin, A.; Gillis, J.A. Is it time to change the reference genome? Genome Biol. 2019, 20, 1–9. [Google Scholar] [CrossRef]
- Porubsky, D.; Ebert, P.; Audano, P.A.; Vollger, M.R.; Harvey, W.T.; Marijon, P.; Ebler, J.; Munson, K.M.; Sorensen, M.; Sulovari, A.; et al. Fully phased human genome assembly without parental data using single-cell strand sequencing and long reads. Nat. Biotechnol. 2021, 39, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Stancu, M.C.; van Roosmalen, M.J.; Renkens, I.; Nieboer, M.M.; Middelkamp, S.; de Ligt, J.; Pregno, G.; Giachino, D.; Mandrile, G.; Valle-Inclan, J.E.; et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Tham, C.Y.; Tirado-Magallanes, R.; Goh, Y.; Fullwood, M.J.; Koh, B.T.H.; Wang, W.; Ng, C.H.; Chng, W.J.; Thiery, A.; Tenen, D.G.; et al. NanoVar: Accurate characterization of patients’ genomic structural variants using low-depth nanopore sequencing. Genome Biol. 2020, 21, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Lv, S.; Hu, M.; Gao, Z.; He, H.; Ma, Q.; Deng, X.W.; Zhu, Z.; Wang, X. Single-Molecule Sequencing Assists Genome Assembly Improvement and Structural Variation Inference. Mol. Plant 2016, 9, 1085–1087. [Google Scholar] [CrossRef] [PubMed]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Krishnakumar, R.; Sinha, A.; Bird, S.W.; Jayamohan, H.; Edwards, H.S.; Schoeniger, J.S.; Patel, K.D.; Branda, S.S.; Bartsch, M.S. Systematic and stochastic influences on the performance of the MinION nanopore sequencer across a range of nucleotide bias. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Teekakirikul, P.; Zhu, W.; Gabriel, G.C.; Young, C.B.; Williams, K.; Martin, L.J.; Hill, J.C.; Richards, T.; Billaud, M.; Phillippi, J.A.; et al. Common deletion variants causing protocadherin-deficiency contribute to the complex genetics of bicuspid aortic valve and left-sided congenital heart defects. Hum. Genet. Genom. Adv. 2021, in press. [Google Scholar]
- Liu, X.; Yagi, H.; Saeed, S.; Bais, A.S.; Gabriel, G.C.; Chen, Z.; Peterson, K.A.; Li, Y.; Schwartz, M.C.; Reynolds, W.T.; et al. The complex genetics of hypoplastic left heart syndrome. Nat. Genet. 2017, 49, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- Schulkey, C.E.; Regmi, S.D.; Magnan, R.A.; Danzo, M.T.; Luther, H.; Hutchinson, A.K.; Panzer, A.A.; Grady, M.M.; Wilson, D.B.; Jay, P.Y. The maternal-age-associated risk of congenital heart disease is modifiable. Nature 2015, 520, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Hinton, R.B.; Martin, L.J.; Rame-Gowda, S.; Tabangin, M.E.; Cripe, L.H.; Benson, D.W. Hypoplastic left heart syndrome links to chromosomes 10q and 6q and is genetically related to bicuspid aortic valve. J. Am. Coll. Cardiol. 2009, 53, 1065–1071. [Google Scholar] [CrossRef]
- McBride, K.L.; Pignatelli, R.; Lewin, M.; Ho, T.; Fernbach, S.; Menesses, A.; Lam, W.; Leal, S.M.; Kaplan, N.; Schliekelman, P.; et al. Inheritance analysis of congenital left ventricular outflow tract obstruction malformations: Segregation, multiplex relative risk, and heritability. Am. J. Med. Genet. A 2005, 134, 180–186. [Google Scholar] [CrossRef]
- Egbe, A.; Uppu, S.; Lee, S.; Ho, D.; Srivastava, S. Prevalence of associated extracardiac malformations in the congenital heart disease population. Pediatr. Cardiol. 2014, 35, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Kusko, R.; Xiao, W.; Zheng, Y.; Liu, Z.; Xiao, C.; Sakkiah, S.; Guo, W.; Gong, P.; Zhang, C.; et al. Similarities and differences between variants called with human reference genome HG19 or HG38. BMC Bioinform. 2019, 20, 17–29. [Google Scholar] [CrossRef]
- Knowler, W.C.; Williams, R.C.; Pettitt, D.J.; Steinberg, A.G. Gm3;5,13,14 and type 2 diabetes mellitus: An association in American Indians with genetic admixture. Am. J. Hum. Genet. 1988, 43, 520–526. [Google Scholar]
- Zheng, G.; Freidlin, B.; Li, Z.; Gastwirth, J.L. Genomic control for association studies under various genetic models. Biometrics 2005, 61, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.G.; Walker, N.M.; Smyth, D.J.; Pask, R.; Cooper, J.D.; Maier, L.M.; Smink, L.J.; Lam, A.C.; Ovington, N.R.; Stevens, H.E.; et al. Population structure, differential bias and genomic control in a large-scale, case-control association study. Nat. Genet. 2005, 37, 1243–1246. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. dbGaP/Database of Genotypes and Phenotypes. Available online: https://www.ncbi.nlm.nih.gov/gap (accessed on 10 March 2020).
- Mailman, M.D.; Feolo, M.; Jin, Y.; Kimura, M.; Tryka, K.; Bagoutdinov, R.; Hao, L.; Kiang, A.; Paschall, J.; Phan, L.; et al. The NCBI dbGaP database of genotypes and phenotypes. Nat. Genet. 2007, 39, 1181–1186. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Cole, J.W.; Grond-Ginsbach, C. Departure from Hardy Weinberg Equilibrium and Genotyping Error. Front. Genet. 2017, 8, 167. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Wang, K.; Hu, X.; Peng, Y. An analytical comparison of the principal component method and the mixed effects model for association studies in the presence of cryptic relatedness and population stratification. Hum. Hered. 2013, 76, 1–9. [Google Scholar] [CrossRef]
- McCarthy, M.I.; Abecasis, G.R.; Cardon, L.R.; Goldstein, D.B.; Little, J.; Ioannidis, J.P.; Hirschhorn, J.N. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat. Rev. Genet. 2008, 9, 356–369. [Google Scholar] [CrossRef]
- Agopian, A.J.; Goldmuntz, E.; Hakonarson, H.; Sewda, A.; Taylor, D.; Mitchell, L.E.; Pediatric Cardiac Genomics Consortium. Genome-Wide Association Studies and Meta-Analyses for Congenital Heart Defects. Circ. Cardiovasc. Genet. 2017, 10, e001449. [Google Scholar] [CrossRef]
- Horvath, S.; Xu, X.; Laird, N.M. The family based association test method: Strategies for studying general genotype--phenotype associations. Eur. J. Hum. Genet. 2001, 9, 301–306. [Google Scholar] [CrossRef]
- Lewinger, J.P.; Bull, S.B. Validity, efficiency, and robustness of a family-based test of association. Genet. Epidemiol. 2006, 30, 62–76. [Google Scholar] [CrossRef]
- Hecker, J.; Townes, F.W.; Kachroo, P.; Laurie, C.; Lasky-Su, J.; Ziniti, J.; Cho, M.H.; Weiss, S.T.; Laird, N.M.; Lange, C. A unifying framework for rare variant association testing in family-based designs, including higher criticism approaches, SKATs, and burden tests. Bioinformatics 2020. [Google Scholar] [CrossRef]
- Zhou, J.J.; Yip, W.K.; Cho, M.H.; Qiao, D.; McDonald, M.L.; Laird, N.M. A comparative analysis of family-based and population-based association tests using whole genome sequence data. BMC Proc. 2014, 8, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.C.; Lee, S.; Cai, T.; Li, Y.; Boehnke, M.; Lin, X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 2011, 89, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Blue, G.M.; Ip, E.; Walker, K.; Kirk, E.P.; Loughran-Fowlds, A.; Sholler, G.F.; Dunwoodie, S.L.; Harvey, R.P.; Giannoulatou, E.; Badawi, N.; et al. Genetic burden and associations with adverse neurodevelopment in neonates with congenital heart disease. Am. Heart J. 2018, 201, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Izarzugaza, J.M.G.; Ellesoe, S.G.; Doganli, C.; Ehlers, N.S.; Dalgaard, M.D.; Audain, E.; Dombrowsky, G.; Banasik, K.; Sifrim, A.; Wilsdon, A.; et al. Systems genetics analysis identifies calcium-signaling defects as novel cause of congenital heart disease. Genome Med. 2020, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Basile, A.O.; Pendergrass, S.A.; Ritchie, M.D. Real world scenarios in rare variant association analysis: The impact of imbalance and sample size on the power in silico. BMC Bioinform. 2019, 20, 1–10. [Google Scholar] [CrossRef]
- Samocha, K.E.; Robinson, E.B.; Sanders, S.J.; Stevens, C.; Sabo, A.; McGrath, L.M.; Kosmicki, J.A.; Rehnstrom, K.; Mallick, S.; Kirby, A.; et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 2014, 46, 944–950. [Google Scholar] [CrossRef]
- Homsy, J.; Zaidi, S.; Shen, Y.; Ware, J.S.; Samocha, K.E.; Karczewski, K.J.; DePalma, S.R.; McKean, D.; Wakimoto, H.; Gorham, J.; et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 2015, 350, 1262–1266. [Google Scholar] [CrossRef]
- Sifrim, A.; Hitz, M.P.; Wilsdon, A.; Breckpot, J.; Turki, S.H.; Thienpont, B.; McRae, J.; Fitzgerald, T.W.; Singh, T.; Swaminathan, G.J.; et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet. 2016, 48, 1060–1065. [Google Scholar] [CrossRef]
- Mathias, R.S.; Lacro, R.V.; Jones, K.L. X-linked laterality sequence: Situs inversus, complex cardiac defects, splenic defects. Am. J. Med. Genet. 1987, 28, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.; Curtis, A.; Korenberg, J.R.; Schipper, R.D.; Allan, L.; Chenevix-Trench, G.; Stephenson, A.; Goodship, J.; Burn, J. A large, dominant pedigree of atrioventricular septal defect (AVSD): Exclusion from the Down syndrome critical region on chromosome 21. Am. J. Hum. Genet. 1993, 53, 1262–1268. [Google Scholar]
- Terrett, J.A.; Newbury-Ecob, R.; Cross, G.S.; Fenton, I.; Raeburn, J.A.; Young, I.D.; Brook, J.D. Holt-Oram syndrome is a genetically heterogeneous disease with one locus mapping to human chromosome 12q. Nat. Genet. 1994, 6, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Satoda, M.; Pierpont, M.E.; Diaz, G.A.; Bornemeier, R.A.; Gelb, B.D. Char syndrome, an inherited disorder with patent ductus arteriosus, maps to chromosome 6p12-p21. Circulation 1999, 99, 3036–3042. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Ramachandran, V.; Cripe, L.H.; Hinton, R.B.; Andelfinger, G.; Tabangin, M.; Shooner, K.; Keddache, M.; Benson, D.W. Evidence in favor of linkage to human chromosomal regions 18q, 5q and 13q for bicuspid aortic valve and associated cardiovascular malformations. Hum. Genet. 2007, 121, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.B.; Buckingham, K.J.; Lee, C.; Bigham, A.W.; Tabor, H.K.; Dent, K.M.; Huff, C.D.; Shannon, P.T.; Jabs, E.W.; Nickerson, D.A.; et al. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010, 42, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alfoldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A structural variation reference for medical and population genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Exome Aggregation, Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jhong, J.H.; Lee, J.; Koo, J.Y. Meta-analytic support vector machine for integrating multiple omics data. BioData Min. 2017, 10, 1–14. [Google Scholar] [CrossRef]
- Miosge, L.A.; Field, M.A.; Sontani, Y.; Cho, V.; Johnson, S.; Palkova, A.; Balakishnan, B.; Liang, R.; Zhang, Y.; Lyon, S.; et al. Andrews, Comparison of predicted and actual consequences of missense mutations. Proc. Natl. Acad. Sci. USA 2015, 112, E5189–E5198. [Google Scholar] [CrossRef]
- Ernst, C.; Hahnen, E.; Engel, C.; Nothnagel, M.; Weber, J.; Schmutzler, R.K.; Hauke, J. Performance of in silico prediction tools for the classification of rare BRCA1/2 missense variants in clinical diagnostics. BMC Med. Genom. 2018, 11, 1–10. [Google Scholar] [CrossRef]
- Williams, K.; Carson, J.; Lo, C. Genetics of Congenital Heart Disease. Biomolecules 2019, 9, 879. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, K.S.; Loffredo, C.A. A cluster of hypoplastic left heart malformation in Baltimore, Maryland. Pediatr. Cardiol. 2006, 27, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, K.J.; Correa, A.; Feinstein, J.A.; Botto, L.; Britt, A.E.; Daniels, S.R.; Elixson, M.; Warnes, C.A.; Webb, C.L. Noninherited risk factors and congenital cardiovascular defects: Current knowledge: A scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: Endorsed by the American Academy of Pediatrics. Circulation 2007, 115, 2995–3014. [Google Scholar] [CrossRef] [PubMed]
- Strickland, M.J.; Klein, M.; Correa, A.; Reller, M.D.; Mahle, W.T.; Riehle-Colarusso, T.J.; Botto, L.D.; Flanders, W.D.; Mulholland, J.A.; Siffel, C.; et al. Ambient air pollution and cardiovascular malformations in Atlanta, Georgia, 1986–2003. Am. J. Epidemiol. 2009, 169, 1004–1014. [Google Scholar] [CrossRef]
- Colliva, A.; Braga, L.; Giacca, M.; Zacchigna, S. Endothelial cell-cardiomyocyte crosstalk in heart development and disease. J. Physiol. 2020, 598, 2923–2939. [Google Scholar] [CrossRef] [PubMed]
- Alkan, F.; Wenzel, A.; Anthon, C.; Havgaard, J.H.; Gorodkin, J. CRISPR-Cas9 off-targeting assessment with nucleic acid duplex energy parameters. Genome Biol. 2018, 19, 1–13. [Google Scholar] [CrossRef]
- Berthelot, C.; Villar, D.; Horvath, J.E.; Odom, D.T.; Flicek, P. Complexity and conservation of regulatory landscapes underlie evolutionary resilience of mammalian gene expression. Nat. Ecol. Evol. 2018, 2, 152–163. [Google Scholar] [CrossRef] [PubMed]
- van Nisselrooij, A.E.L.; Lugthart, M.A.; Clur, S.A.; Linskens, I.H.; Pajkrt, E.; Rammeloo, L.A.; Rozendaal, L.; Blom, N.A.; van Lith, J.M.M.; Knegt, A.C.; et al. The prevalence of genetic diagnoses in fetuses with severe congenital heart defects. Genet. Med. 2020, 22, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Chaix, M.A.; Andelfinger, G.; Khairy, P. Genetic testing in congenital heart disease: A clinical approach. World J. Cardiol. 2016, 8, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Sud, A.; Turnbull, C.; Houlston, R. Will polygenic risk scores for cancer ever be clinically useful? NPJ Precis. Oncol. 2021, 5, 1–5. [Google Scholar] [CrossRef]
- Lewis, A.C.F.; Green, R.C. Polygenic risk scores in the clinic: New perspectives needed on familiar ethical issues. Genome Med. 2021, 13, 1–10. [Google Scholar] [CrossRef]

| Phenomenon | Attribute |
|---|---|
| Genetic heterogeneity | Similar phenotypes, different genetic cause. |
| Variable expressivity | Individuals with same disease gene have different phenotypes. |
| Reduced penetrance | Disease absence in some individuals with disease gene. |
| Pleiotropy | Multiple phenotypes associated with the same genetic cause. |
| Study Design | Case Type | Control Type | Analysis Type | Limitations |
|---|---|---|---|---|
| Case Control | Unrelated | Unrelated -local | Association | Heterogeneity |
| Unrelated—Out of study | Association | Heterogeneity Differences between cases and controls Differences in genotype generation | ||
| Trio | Unrelated | Parents of cases | Family Based Association (TDT) | Heterogeneity |
| Linkage analyses | Heterogeneity | |||
| Filtering | Generalizability | |||
| Family | Related—may be a series of families | Family members of cases | Family based Association | Heterogeneity |
| Linkage analysis | Generalizability | |||
| Filtering | Generalizability |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, L.J.; Benson, D.W. Focused Strategies for Defining the Genetic Architecture of Congenital Heart Defects. Genes 2021, 12, 827. https://doi.org/10.3390/genes12060827
Martin LJ, Benson DW. Focused Strategies for Defining the Genetic Architecture of Congenital Heart Defects. Genes. 2021; 12(6):827. https://doi.org/10.3390/genes12060827
Chicago/Turabian StyleMartin, Lisa J., and D. Woodrow Benson. 2021. "Focused Strategies for Defining the Genetic Architecture of Congenital Heart Defects" Genes 12, no. 6: 827. https://doi.org/10.3390/genes12060827
APA StyleMartin, L. J., & Benson, D. W. (2021). Focused Strategies for Defining the Genetic Architecture of Congenital Heart Defects. Genes, 12(6), 827. https://doi.org/10.3390/genes12060827