
Mutational Asymmetries in the SARS-CoV-2 Genome May Lead to Increased Hydrophobicity of Virus Proteins



Abstract
1. Introduction
2. Materials and Methods
2.1. Source of Sequences
2.2. Analysis of Variants
2.3. Phylogenetic Analysis
2.4. Protein Analysis
2.5. Data Processing
3. Results
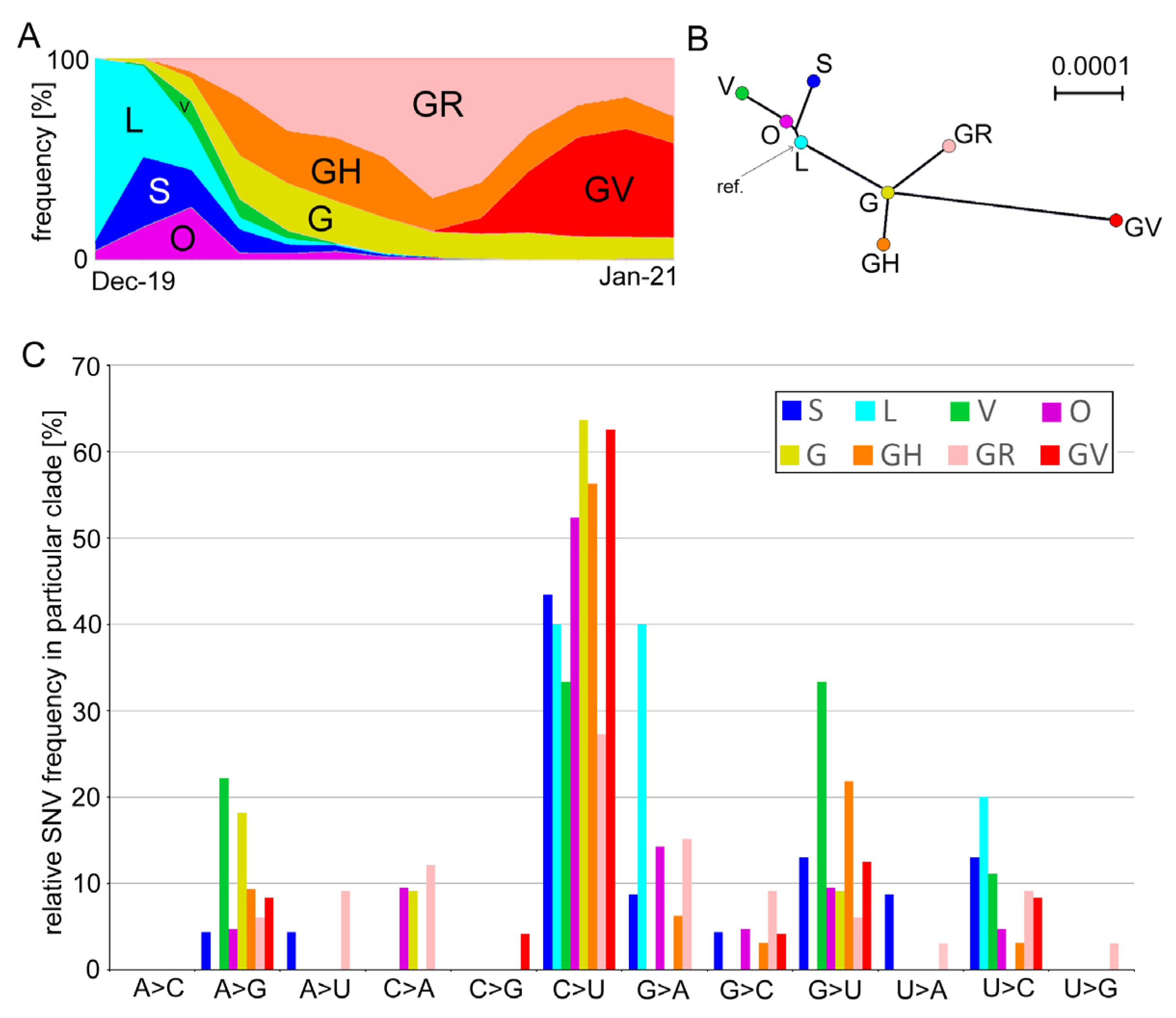
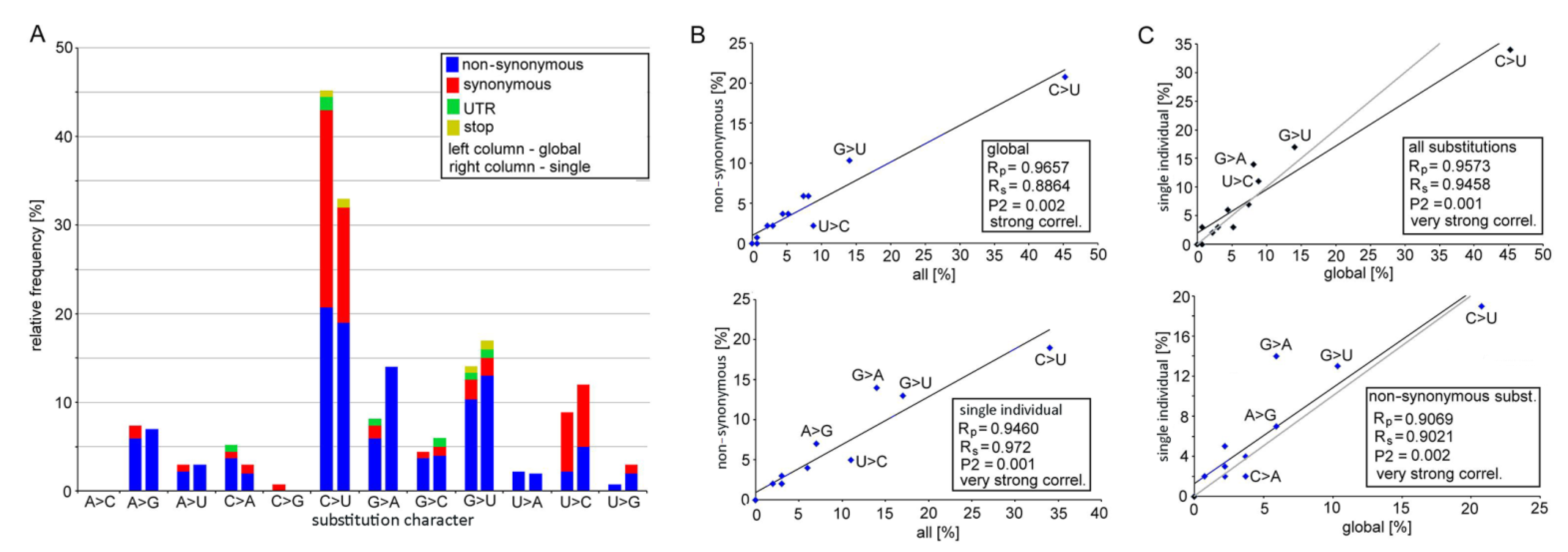
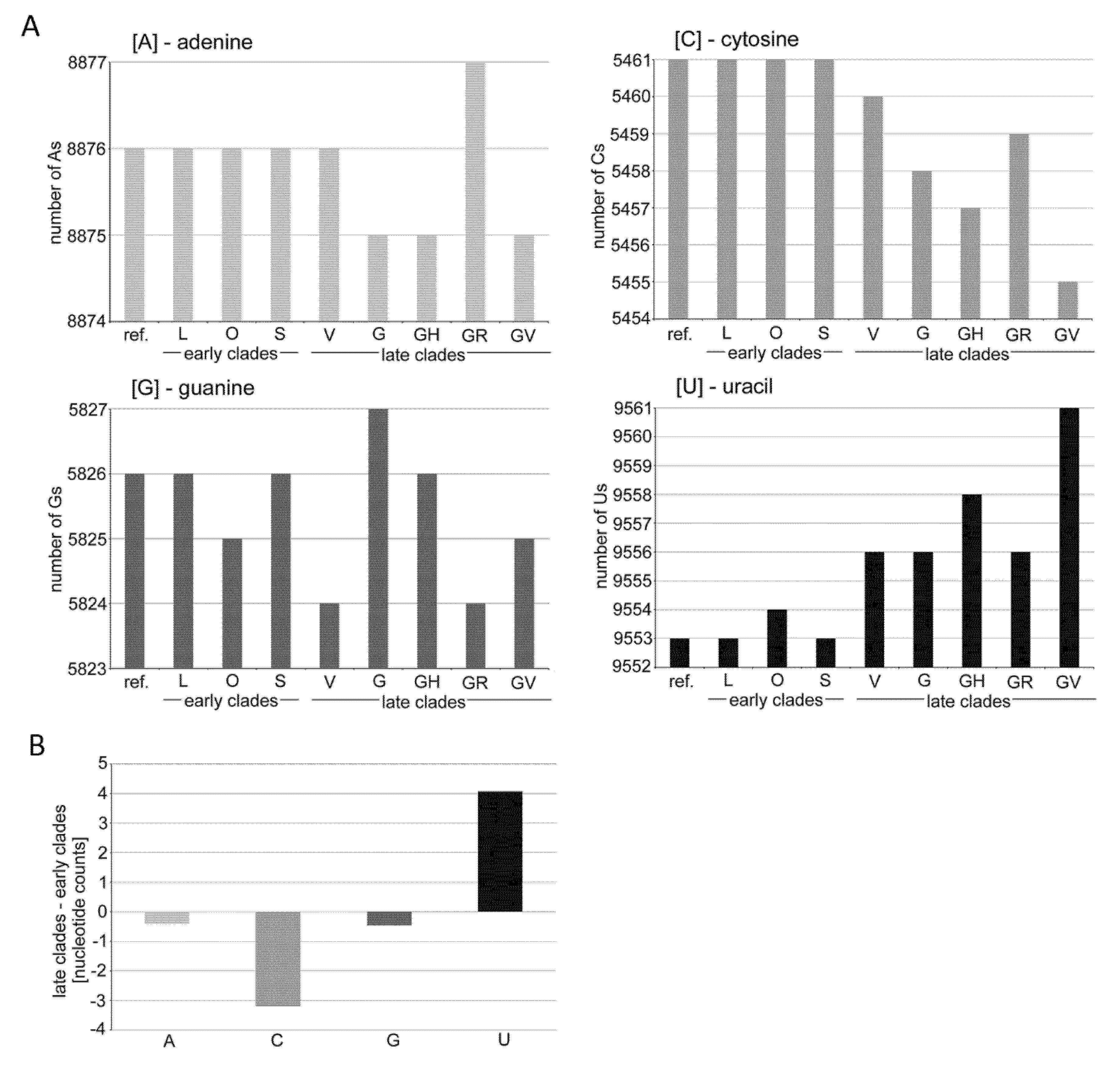
3.1. Mutation Spectra in the SARS-CoV-2 Clades
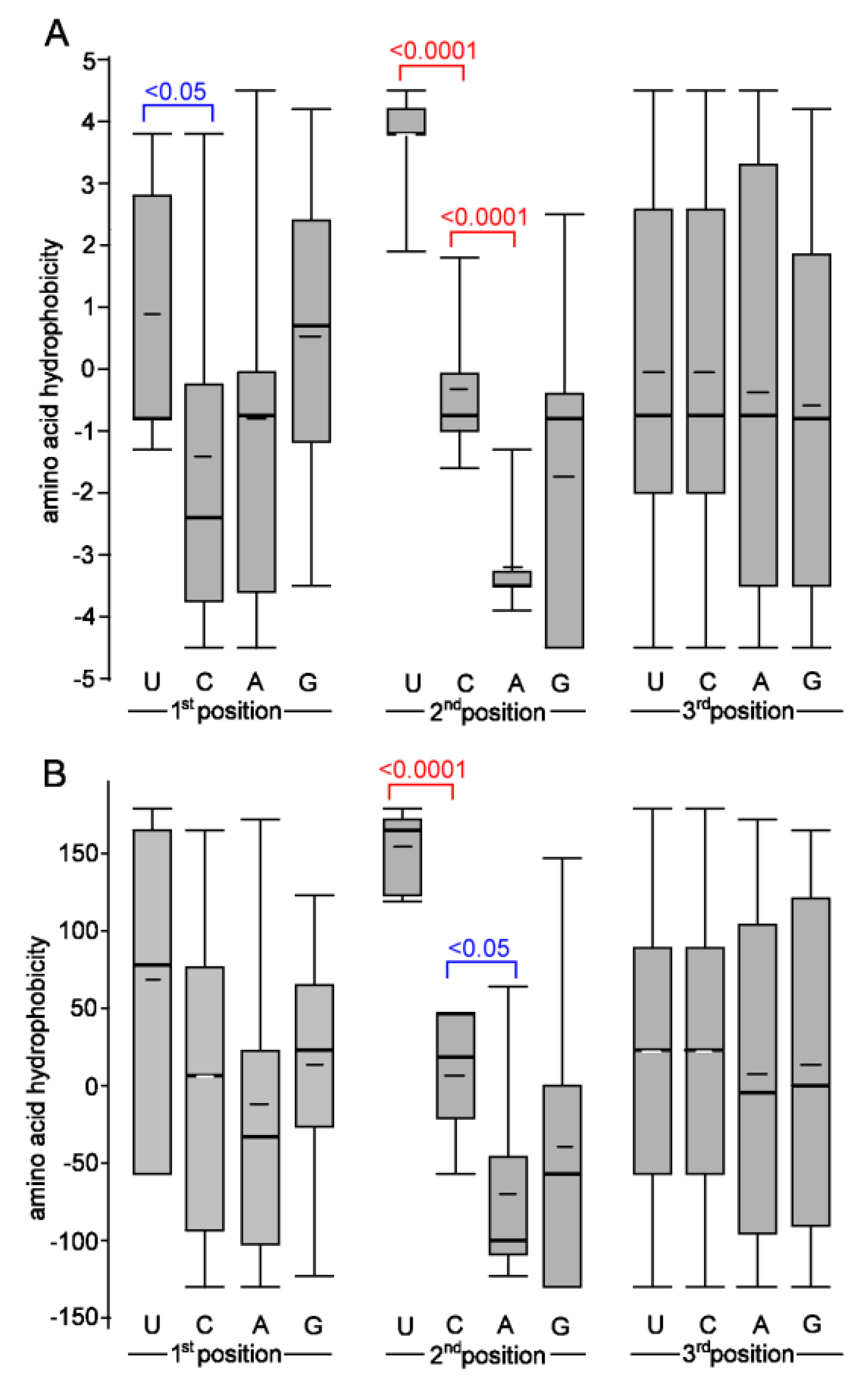
3.2. Biochemical Properties of Amino Acids Changed by Nucleotide Substitutions
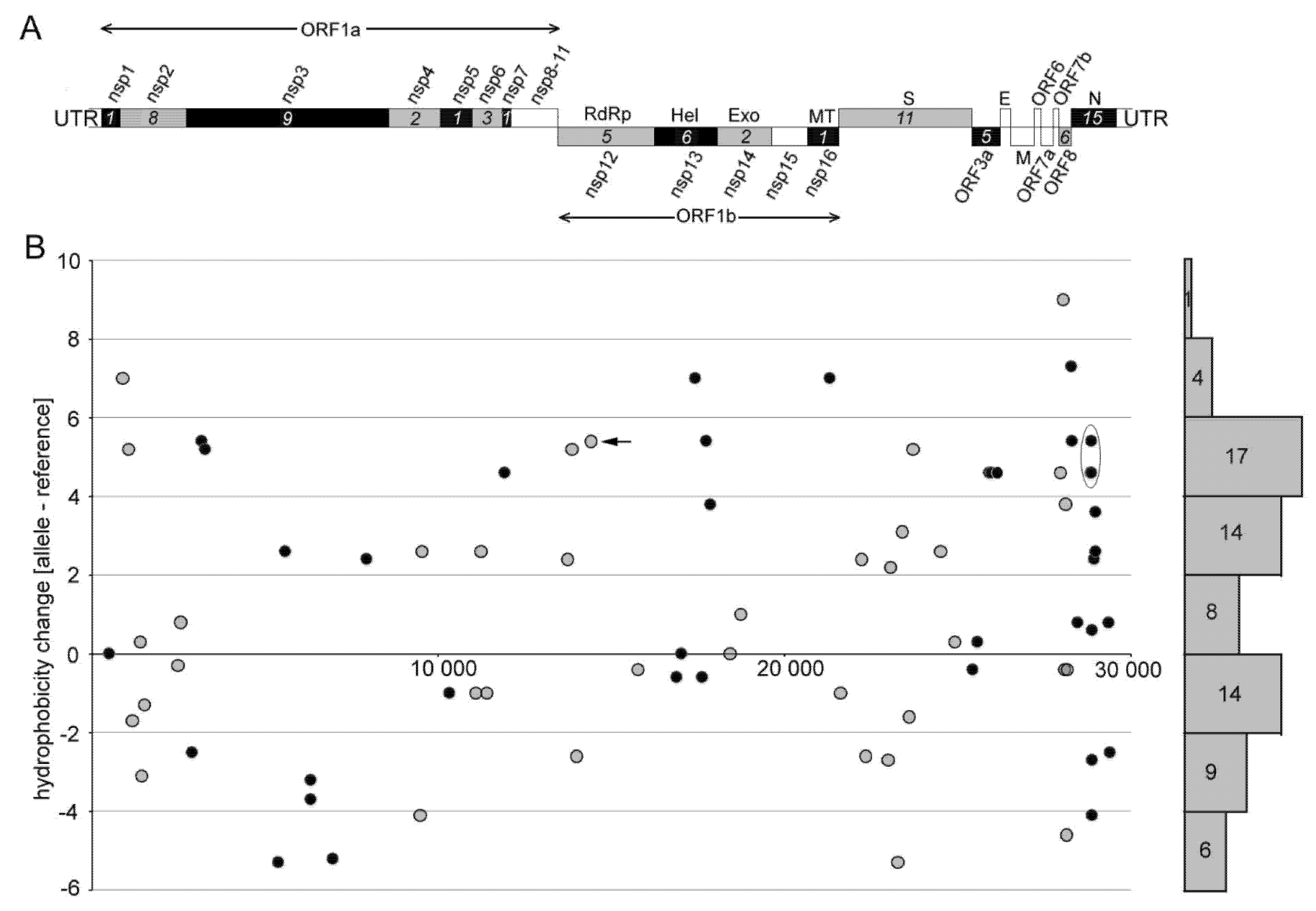
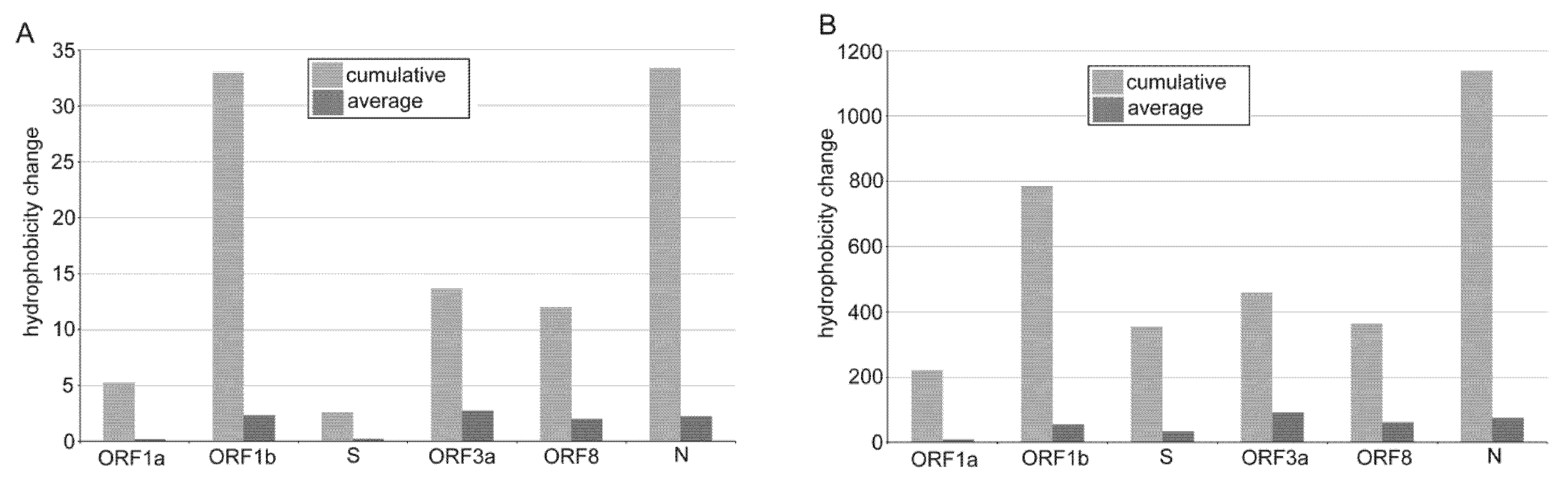
3.3. The Impact of Emerging Mutations on Virus Protein Domains
4. Discussion
4.1. Possible Mechanisms Leading to Mutation Asymmetries in the SARS-CoV-2 Genomes
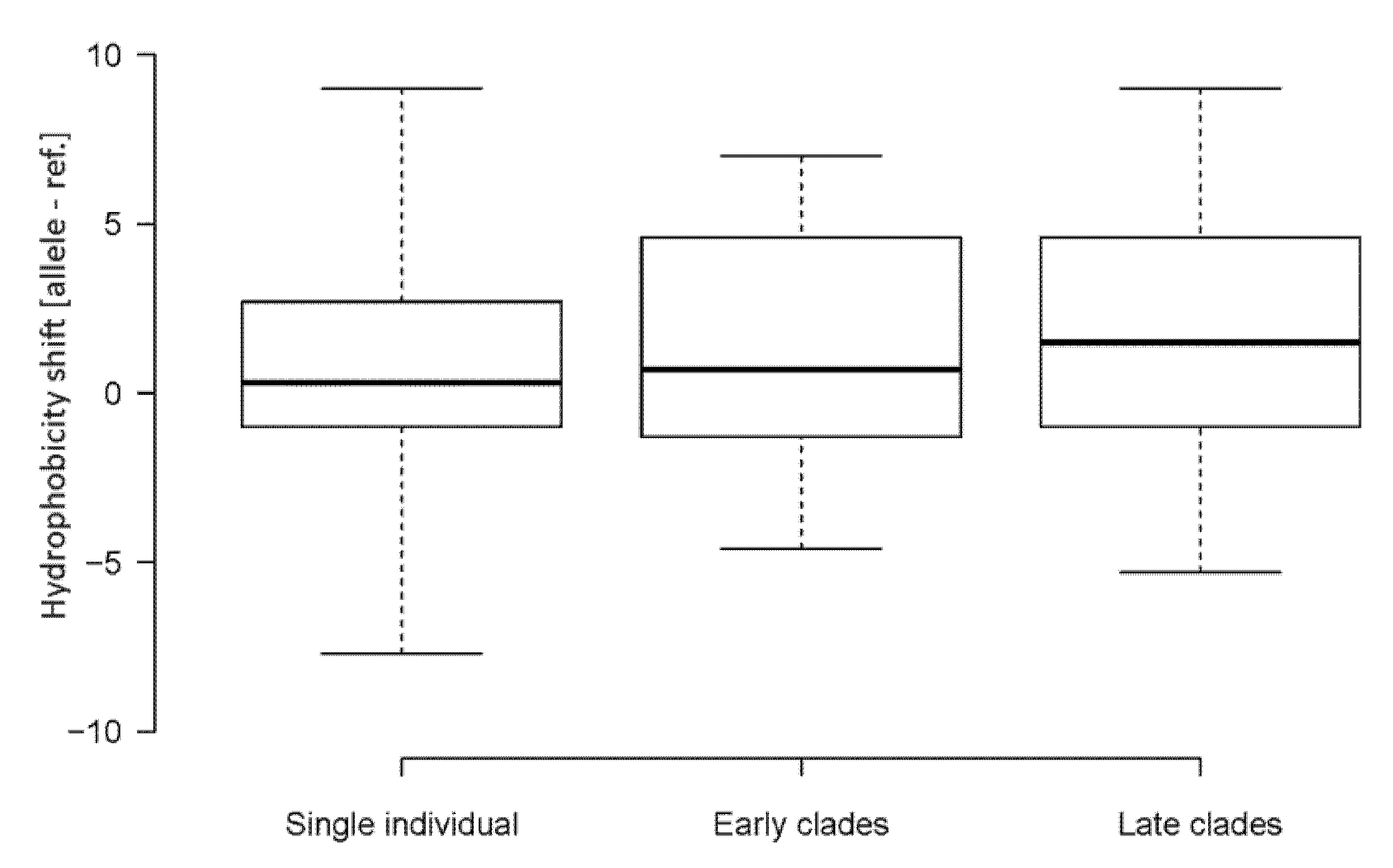
4.2. Emerging Nucleotide Substitutions May Increase the Hydrophobicity of Viral Proteins
4.3. Relationship between the Nucleotide Composition of Codons and Amino Acid Hydrophobicity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duffy, S. Why are RNA virus mutation rates so damn high? PLoS Biol. 2018, 16, e3000003. [Google Scholar] [CrossRef] [PubMed]
- Ratcliff, J.; Simmonds, P. Potential APOBEC-mediated RNA editing of the genomes of SARS-CoV-2 and other coronaviruses and its impact on their longer term evolution. Virology 2021, 556, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Roy, C.; Mandal, S.M.; Mondal, S.K.; Mukherjee, S.; Mapder, T.; Ghosh, W.; Chakraborty, R. Trends of mutation accumulation across global SARS-CoV-2 genomes: Implications for the evolution of the novel coronavirus. Genomics 2020, 112, 5331–5342. [Google Scholar] [CrossRef] [PubMed]
- Koyama, T.; Platt, D.; Parida, L. Variant analysis of SARS-CoV-2 genomes. Bull. World Health Organ. 2020, 98, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Phan, T. Genetic diversity and evolution of SARS-CoV-2. Infect. Genet. Evol. 2020, 81, 104260. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.F.; Wang, L.P.; Fang, Y.; Wang, L.Y. Global SNP analysis of 11,183 SARS-CoV-2 strains reveals high genetic diversity. Transbound. Emerg. Dis. 2020. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Holmes, E.C. A Genomic Perspective on the Origin and Emergence of SARS-CoV-2. Cell 2020, 181, 223–227. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Boni, M.F.; Lemey, P.; Jiang, X.W.; Lam, T.T.Y.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef]
- Li, X.; Giorgi, E.E.; Marichannegowda, M.H.; Foley, B.; Xiao, C.; Kong, X.P.; Chen, Y.; Gnanakaran, S.; Korber, B.; Gao, F. Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci. Adv. 2020, 6, eabb9153. [Google Scholar] [CrossRef]
- Frutos, R.; Gavote, L.; Devaux, C. Understanding the origin of COVID-19 requires to change the paradigm on zoonotic emergence from the spillover model to the circulation model. Infect. Genet. Evol. 2021, 104812. [Google Scholar] [CrossRef] [PubMed]
- Vijgen, L.; Keyaerts, E.; Moes, E.; Thoelen, I.; Wollants, E.; Lemey, P.; Vandamme, A.M.; Van Ranst, M. Complete genomic sequence of human coronavirus OC43: Molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J. Virol. 2005, 79, 1595–1604. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, H.; Wu, X.; Zhong, Y.; Zhang, K.; Zhang, Y.P.; Boerwinkle, E.; Fu, Y.X. Moderate mutation rate in the SARS coronavirus genome and its implications. BMC Evol. Biol. 2004, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- van Dorp, L.; Acman, M.; Richard, D.; Shaw, L.P.; Ford, C.E.; Ormond, L.; Owen, C.J.; Pang, J.; Tan, C.C.S.; Boshier, F.A.T.; et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. Evol. 2020, 83, 104351. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Pipes, L.; Nielsen, R. Synonymous mutations and the molecular evolution of SARS-CoV-2 origins. Virus Evol. 2021, 7, veaa098. [Google Scholar] [CrossRef]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75.e11. [Google Scholar] [CrossRef]
- Dearlove, B.; Lewitus, E.; Bai, H.; Li, Y.; Reeves, D.B.; Joyce, M.G.; Scott, P.T.; Amare, M.F.; Vasan, S.; Michael, N.L.; et al. A SARS-CoV-2 vaccine candidate would likely match all currently circulating variants. Proc. Natl. Acad. Sci. USA 2020, 117, 23652–23662. [Google Scholar] [CrossRef]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, K.; Steininger, P.; Ziegler, R.; Steinmann, J.; Korn, K.; Ensser, A. SARS-CoV-2 samples may escape detection because of a single point mutation in the N gene. Eurosurveillance 2020, 25, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Matyášek, R.; Kovařík, A. Mutation Patterns of Human SARS-CoV-2 and Bat RaTG13 Coronavirus Genomes Are Strongly Biased Towards C>U Transitions, Indicating Rapid Evolution in Their Hosts. Genes 2020, 11, 761. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P. Rampant C-->U Hypermutation in the Genomes of SARS-CoV-2 and Other Coronaviruses: Causes and Consequences for Their Short- and Long-Term Evolutionary Trajectories. mSphere 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Vankadari, N. Overwhelming mutations or SNPs of SARS-CoV-2: A point of caution. Gene 2020, 752, 144792. [Google Scholar] [CrossRef] [PubMed]
- Klimczak, L.J.; Randall, T.A.; Saini, N.; Li, J.L.; Gordenin, D.A. Similarity between mutation spectra in hypermutated genomes of rubella virus and in SARS-CoV-2 genomes accumulated during the COVID-19 pandemic. PLoS ONE 2020, 15, e0237689. [Google Scholar] [CrossRef]
- Mourier, T.; Sadykov, M.; Carr, M.J.; Gonzalez, G.; Hall, W.W.; Pain, A. Host-directed editing of the SARS-CoV-2 genome. Biochem. Biophys. Res. Commun. 2020, 538, 35–39. [Google Scholar] [CrossRef]
- Nabel, C.S.; Manning, S.A.; Kohli, R.M. The Curious Chemical Biology of Cytosine: Deamination, Methylation, and Oxidation as Modulators of Genomic Potential. ACS Chem. Biol. 2012, 7, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Kandeel, M.; Ibrahim, A.; Fayez, M.; Al-Nazawi, M. From SARS and MERS CoVs to SARS-CoV-2: Moving toward more biased codon usage in viral structural and non-structural genes. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Nyayanit, D.A.; Yadav, P.D.; Kharde, R.; Cherian, S. Natural Selection Plays an Important Role in Shaping the Codon Usage of Structural Genes of the Viruses Belonging to the Coronaviridae Family. Viruses 2021, 13, 3. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef]
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; Ferreira, I.A.T.M.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Lumb, I.U.; et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 2021, 592, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Palecz, B. Enthalpic homogeneous pair interaction coefficients of L-alpha-amino acids as a hydrophobicity parameter of amino acid side chains. J. Am. Chem. Soc. 2002, 124, 6003–6008. [Google Scholar] [CrossRef] [PubMed]
- Welling, G.W.; Weijer, W.J.; Vanderzee, R.; Wellingwester, S. Prediction of Sequential Antigenic Regions in Proteins. FEBS Lett. 1985, 188, 215–218. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; RStudio and Inc. Shiny: Web Application Framework for R. R Package Version 0.5.0; R Foundation for Statistical Computing: Vienna, Austria, 2013; Available online: http://shiny.chemgrid.org/boxplotr/ (accessed on 15 February 2021).
- Mann Whitney U Test Calculator. Statistics Kingdom. 2017. Available online: https://www.statskingdom.com/about.html (accessed on 6 May 2021).
- Tang, X.; Wu, C.; Li, X.; Song, Y.; Yao, X.; Wu, X.; Duan, Y.; Zhang, H.; Wang, Y.; Qian, Z.; et al. On the origin and continuing evolution of SARS-CoV-2. Natl. Sci. Rev. 2020, 7, 1012–1023. [Google Scholar] [CrossRef]
- Troyano-Hernaez, P.; Reinosa, R.; Holguin, A. Evolution of SARS-CoV-2 Envelope, Membrane, Nucleocapsid, and Spike Structural Proteins from the Beginning of the Pandemic to September 2020: A Global and Regional Approach by Epidemiological Week. Viruses 2021, 13, 243. [Google Scholar] [CrossRef]
- SARS-CoV-2 Variants of Concern. Available online: https://www.ecdc.europa.eu/en/covid-19/variants-concern (accessed on 6 May 2021).
- IDSA Contributor. COVID Mega-Variant and Eight Criteria for a Template to Assess All Variants. Science Speaks: Global ID News. Available online: https://sciencespeaksblog.org/2021/02/02/covid-mega-variant-and-eight-criteria-for-a-template-to-assess-all-variants/ (accessed on 10 March 2021).
- Bhattacharjee, S. COVID-19|A.P. Strain at Least 15 Times more Virulent. Available online: https://www.thehindu.com/news/national/andhra-pradesh/ap-strain-at-least-15-times-more-virulent/article34474035.ece (accessed on 5 May 2021).
- Greenwood, M. What Mutations of SARS-CoV-2 are Causing Concern? Available online: https://www.news-medical.net/health/What-Mutations-of-SARS-CoV-2-are-Causing-Concern.aspx (accessed on 18 March 2021).
- Singh, A.; Steinkellner, G.; Kochl, K.; Gruber, K.; Gruber, C.C. Serine 477 plays a crucial role in the interaction of the SARS-CoV-2 spike protein with the human receptor ACE2. Sci. Rep. 2021, 11, 4320. [Google Scholar] [CrossRef]
- Wise, J. Covid-19: The E484K mutation and the risks it poses. BMJ 2021, 372, n359. [Google Scholar] [CrossRef]
- Shahhosseini, N.; Babuadze, G.; Wong, G.; Kobinger, G. Mutation Signatures and In Silico Docking of Novel SARS-CoV-2 Variants of Concern. Microorganisms 2021, 9, 926. [Google Scholar] [CrossRef]
- Wang, R.; Hozumi, Y.; Zheng, Y.H.; Yin, C.C.; Wei, G.W. Host Immune Response Driving SARS-CoV-2 Evolution. Viruses 2020, 12, 1095. [Google Scholar] [CrossRef]
- Sharma, S.; Patnaik, S.K.; Taggart, R.T.; Kannisto, E.D.; Enriquez, S.M.; Gollnick, P.; Baysal, B.E. APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat. Commun. 2015, 6, 6881. [Google Scholar] [CrossRef]
- Milewska, A.; Kindler, E.; Vkovski, P.; Zeglen, S.; Ochman, M.; Thiel, V.; Rajfur, Z.; Pyrc, K. APOBEC3-mediated restriction of RNA virus replication. Sci. Rep. 2018, 8, 5960. [Google Scholar] [CrossRef]
- Poole, A.; Penny, D.; Sjoberg, B.M. Confounded cytosine! Tinkering and the evolution of DNA. Nat. Rev Mol. Cell Biol. 2001, 2, 147–151. [Google Scholar] [CrossRef]
- Goswami, P.; Bartas, M.; Lexa, M.; Bohalova, N.; Volna, A.; Cerven, J.; Cervenova, V.; Pecinka, P.; Spunda, V.; Fojta, M.; et al. SARS-CoV-2 hot-spot mutations are significantly enriched within inverted repeats and CpG island loci. Brief. Bioinform. 2020, 22, 1338–1345. [Google Scholar] [CrossRef]
- Graudenzi, A.; Maspero, D.; Angaroni, F.; Piazza, R.; Ramazzotti, D. Mutational signatures and heterogeneous host response revealed via large-scale characterization of SARS-CoV-2 genomic diversity. iScience 2021, 24, 102116. [Google Scholar] [CrossRef] [PubMed]
- Garvin, M.R.; Prates, E.T.; Pavicic, M.; Jones, P.; Amos, B.K.; Geiger, A.; Shah, M.B.; Streich, J.; Gazolla, J.G.F.M.; Kainer, D.; et al. Potentially adaptive SARS-CoV-2 mutations discovered with novel spatiotemporal and explainable AI models. Genome Biol. 2020, 21, 304. [Google Scholar] [CrossRef] [PubMed]
- Yarus, M. Crick Wobble and Superwobble in Standard Genetic Code Evolution. J. Mol. Evol. 2021, 89, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Minskaia, E.; Hertzig, T.; Gorbalenya, A.E.; Campanacci, V.; Cambillau, C.; Canard, B.; Ziebuhr, J. Discovery of an RNA virus 3′->5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 5108–5113. [Google Scholar] [CrossRef]
- Velthuis, A.J.W.T.; van den Worm, S.H.E.; Snijder, E.J. The SARS-coronavirus nsp7+nsp8 complex is a unique multimeric RNA polymerase capable of both de novo initiation and primer extension. Nucleic Acids Res. 2012, 40, 1737–1747. [Google Scholar] [CrossRef]
- Jia, Z.H.; Yan, L.M.; Ren, Z.L.; Wu, L.J.; Wang, J.; Guo, J.; Zheng, L.T.; Ming, Z.H.; Zhang, L.Q.; Lou, Z.Y.; et al. Delicate structural coordination of the Severe Acute Respiratory Syndrome coronavirus Nsp13 upon ATP hydrolysis. Nucleic Acids Res. 2019, 47, 6538–6550. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Z.; Liu, C.; Guo, Y.; He, Z.; Huang, X.; Jia, X.; Yang, T. SARS-CoV-2 and SARS-CoV: Virtual screening of potential inhibitors targeting RNA-dependent RNA polymerase activity (NSP12). J. Med. Virol. 2021, 93, 389–400. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Clade | Number of Genomes | Coverage | Characteristic Nucleotide Variations 1 | |
|---|---|---|---|---|---|
| Total 2 | This Study | (%) | |||
| Early | L | 4699 | 2222 | 47.3 | C241, C3037, C8782, G11083, A23403, G25563, and U28144C |
| O | 5681 | 2714 | 47.8 | G11083U, C22227U, A23403G, and G26144U | |
| S | 7893 | 4532 | 57.4 | C8782U and U28144C | |
| V | 5320 | 1896 | 35.6 | C241U, C28311U, and C23929U | |
| Late | G | 62,786 | 8638 | 13.8 | C241U, C3037U, and A23403G |
| GH | 89,908 | 23,375 | 26 | C241U, C3037U, A23403G, and G25563U | |
| GR | 136,083 | 35,857 | 26.3 | C241U, C3037U, A23403G, and A28111G | |
| GV | 92,617 | 15,930 | 17.2 | C241U, C3037U, A23403G, and C22227U | |
| Name 1 | Mutation coordinate | Amino Acid Hydrophobicity 2 | Note | |||
|---|---|---|---|---|---|---|
| Genome Protein | Ref. | Allele | Shift | |||
| N440K | U22882G | Asp440Lys | −3.5 | −3.9 | −0.4 | Suspected to increase the infectivity of the virus [42] |
| L452R | U22917G | Leu452Arg | 3.8 | −4.5 | −8.3 | Thought to increase immune evasion and ACE2 binding [43] |
| S477G * | A22991G | Ser477Gly | −0.8 | −0.4 | 0.4 | Suspected to strengthen receptor interaction [44] |
| S477N | G22992A | Ser477Asp | −0.8 | −3.5 | −2.7 | Strengthens receptor interaction [44] |
| E484K | G23012A | Glu484Lys | −3.5 | −3.9 | −0.4 | Increased evasion from the host’s immune system [45] |
| E484Q | A23014C | Glu484Gln | −3.5 | −3.5 | 0 | Is suspected to increase the infectivity of the virus |
| N501Y | A23063U | Asn501Tyr | −3.5 | −1.3 | 2.2 | Enhances binding activity to the ACE2 receptor and is a variant of concern [46] |
| D614G * | A23604G | Asp614Gly | −3.5 | −0.4 | 3.1 | Dominant form in the pandemic [7] |
| P681H | C23604A | Pro681His | −1.6 | −3.2 | −1.6 | Increasing prevalence worldwide [43] |
| P681R * | C23604G | Pro681Arg | −1.6 | −4.5 | −2.9 | May evade the immune system [43] |
| Total | −18.5 | −29.1 | −10.6 | |||
| Average | −1.85 | −2.91 | −1.06 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matyášek, R.; Řehůřková, K.; Berta Marošiová, K.; Kovařík, A. Mutational Asymmetries in the SARS-CoV-2 Genome May Lead to Increased Hydrophobicity of Virus Proteins. Genes 2021, 12, 826. https://doi.org/10.3390/genes12060826
Matyášek R, Řehůřková K, Berta Marošiová K, Kovařík A. Mutational Asymmetries in the SARS-CoV-2 Genome May Lead to Increased Hydrophobicity of Virus Proteins. Genes. 2021; 12(6):826. https://doi.org/10.3390/genes12060826
Chicago/Turabian StyleMatyášek, Roman, Kateřina Řehůřková, Kristýna Berta Marošiová, and Aleš Kovařík. 2021. "Mutational Asymmetries in the SARS-CoV-2 Genome May Lead to Increased Hydrophobicity of Virus Proteins" Genes 12, no. 6: 826. https://doi.org/10.3390/genes12060826
APA StyleMatyášek, R., Řehůřková, K., Berta Marošiová, K., & Kovařík, A. (2021). Mutational Asymmetries in the SARS-CoV-2 Genome May Lead to Increased Hydrophobicity of Virus Proteins. Genes, 12(6), 826. https://doi.org/10.3390/genes12060826