
The TNFRSF13C H159Y Variant Is Associated with Severe COVID-19: A Retrospective Study of 500 Patients from Southern Italy

 ,
,  ,
, 
 , , , , ,
, , , , ,  ,
,  and
and  add
Show full author list
add
Show full author list
Abstract
1. Introduction
2. Materials and Methods
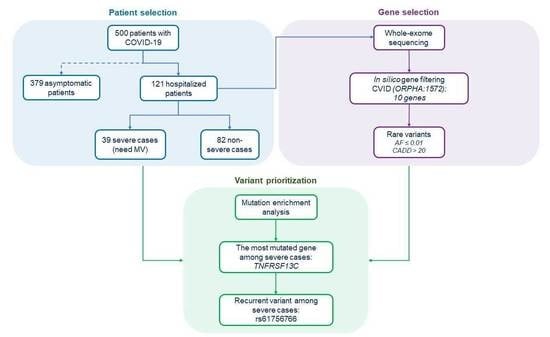
2.1. Collection and Clinical Stratification of the Patients
2.2. DNA Extraction, Quantification, and Library Preparation for Sequencing
2.3. Bioinformatic Analysis of Sequencing Data
2.4. Selection of CVID-Related Genes
2.5. SNP Genotyping
3. Results
3.1. Mutational Enrichment Analysis
3.2. Identification of a Recurrent TNFRSF13C Variant Associated with COVID-19 Severe Phenotype
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anastassopoulou, C.; Gkizarioti, Z.; Patrinos, G.P.; Tsakris, A. Human genetic factors associated with susceptibility to SARS-CoV-2 infection and COVID-19 disease severity. Hum. Genom. 2020, 14, 40. [Google Scholar] [CrossRef]
- Elhabyan, A.; Elyaacoub, S.; Sanad, E.; Abukhadra, A.; Elhabyan, A.; Dinu, V. The role of host genetics in susceptibility to severe viral infections in humans and insights into host genetics of severe COVID-19: A systematic review. Virus Res. 2020, 289, 198163. [Google Scholar] [CrossRef]
- Benetti, E.; Tita, R.; Spiga, O.; Ciolfi, A.; Birolo, G.; Bruselles, A.; Doddato, G.; Giliberti, A.; Marconi, C.; Musacchia, F.; et al. ACE2 gene variants may underlie interindividual variability and susceptibility to COVID-19 in the Italian population. Eur. J. Hum. Genet. 2020, 8, 1602–1614. [Google Scholar] [CrossRef] [PubMed]
- Russo, R.; Andolfo, I.; Lasorsa, V.A.; Iolascon, A.; Capasso, M. Genetic Analysis of the Coronavirus SARS-CoV-2 Host Protease TMPRSS2 in Different Populations. Front. Genet. 2020, 11, 872. [Google Scholar] [CrossRef] [PubMed]
- Asselta, R.; Paraboschi, E.M.; Mantovani, A.; Duga, S. ACE2 and TMPRSS2 variants and expression as candidates to sex and country differences in COVID-19 severity in Italy. Aging 2020, 12, 10087–10098. [Google Scholar] [CrossRef] [PubMed]
- Andolfo, I.; Russo, R.; Lasorsa, V.A.; Cantalupo, S.; Rosato, B.E.; Bonfiglio, F.; Frisso, G.; Abete, P.; Cassese, G.M.; Servillo, G.; et al. Common variants at 21q22.3 locus influence MX1 and TMPRSS2 gene expression and susceptibility to severe COVID-19. iScience 2021, 24, 102322. [Google Scholar] [CrossRef]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92–98. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef]
- Severe Covid-19 GWAS Group; Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernández, J.; Prati, D.; Baselli, G.; et al. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar] [PubMed]
- Carter-Timofte, M.E.; Jørgensen, S.E.; Freytag, M.R.; Thomsen, M.M.; Andersen, N.S.B.; Al-Mousawi, A.; Hait, A.S.; Mogensen, T.H. Deciphering the Role of Host Genetics in Susceptibility to Severe COVID-19. Front. Immunol. 2020, 11, 1606. [Google Scholar] [CrossRef]
- Quinti, I.; Lougaris, V.; Milito, C.; Cinetto, F.; Pecoraro, A.; Mezzaroma, I.; Mastroianni, C.M.; Turriziani, O.; Bondioni, M.P.; Filippini, M.; et al. A possible role for B cells in COVID-19? Lesson from patients with agammaglobulinemia. J. Allergy Clin. Immunol. 2020, 146, 211–213. [Google Scholar] [CrossRef]
- Mackay, F.; Schneider, P. Cracking the BAFF code. Nat. Rev. Immunol. 2009, 9, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wang, Y.; Shao, C.; Huang, J.; Gan, J.; Huang, X.; Bucci, E.; Piacentini, M.; Ippolito, G.; Melino, G. COVID-19 infection: The perspectives on immune responses. Cell Death Differ. 2020, 27, 1451–1454. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, J.M.; Luo, Z.; Manske, M.K.; Price-Troska, T.; Ziesmer, S.C.; Lin, W.; Hostager, B.S.; Slager, S.L.; Witzig, T.E.; Ansell, S.M.; et al. A BAFF-R mutation associated with non-Hodgkin lymphoma alters TRAF recruitment and reveals new insights into BAFF-R signaling. J. Exp. Med. 2010, 207, 2569–2579. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, A.; Mavragani, C.P.; Nezos, A.; Zintzaras, E.; Quartuccio, L.; De Vita, S.; Koutsilieris, M.; Tzioufas, A.G.; Moutsopoulos, H.M.; Voulgarelis, M. A BAFF receptor His159Tyr mutation in Sjogren’s syndrome-related lymphoproliferation. Arthritis Rheumatol. 2015, 67, 2732–2741. [Google Scholar] [CrossRef] [PubMed]
- Lougaris, V.; Vitali, M.; Baronio, M.; Tampella, G.; Plebani, A. BAFF-R mutations in Good’s syndrome. Clin. Immunol. 2014, 153, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Miano, M.; Cappelli, E.; Pezzulla, A.; Venè, R.; Grossi, A.; Terranova, P.; Palmisani, E.; Maggiore, R.; Guardo, D.; Lanza, T.; et al. FAS-mediated apoptosis impairment in patients with ALPS/ALPS-like phenotype carrying variants on CASP10 gene. Br. J. Haematol. 2019, 187, 502–508. [Google Scholar] [CrossRef]
- Lougaris, V.; Baronio, M.; Moratto, D.; Cardinale, F.; Plebani, A. Monoallelic BAFFR P21R/H159Y Mutations and Familiar Primary Antibody Deficiencies. J. Clin. Immunol. 2016, 36, 1–3. [Google Scholar] [CrossRef]
- Kutukculer, N.; Gulez, N.; Karaca, N.E.; Aksu, G.; Berdeli, A. Three different classifications, B lymphocyte subpopulations, TNFRSF13B (TACI), TNFRSF13C (BAFF-R), TNFSF13 (APRIL) gene mutations, CTLA-4 and ICOS gene polymorphisms in Turkish patients with common variable immunodeficiency. J. Clin. Immunol. 2012, 32, 1165–1179. [Google Scholar] [CrossRef]
- Ntellas, P.; Dardiotis, E.; Sevdali, E.; Siokas, V.; Aloizou, A.M.; Tsinti, G.; Germenis, A.E.; Hadjigeorgiou, G.M.; Eibel, H.; Speletas, M. TNFRSF13C/BAFFR P21R and H159Y polymorphisms in multiple sclerosis. Mult. Scler. Relat. Disord. 2020, 37, 101422. [Google Scholar] [CrossRef]
- Jasek, M.; Bojarska-Junak, A.; Wagner, M.; Sobczyński, M.; Wołowiec, D.; Roliński, J.; Karabon, L.; Kuśnierczyk, P. Association of variants in BAFF (rs9514828 and rs1041569) and BAFF-R (rs61756766) genes with the risk of chronic lymphocytic leukemia. Tumour. Biol. 2016, 37, 13617–13626. [Google Scholar] [CrossRef] [PubMed]
- Losi, C.G.; Silini, A.; Fiorini, C.; Soresina, A.; Meini, A.; Ferrari, S.; Notarangelo, L.D.; Lougaris, V.; Plebani, A. Mutational analysis of human BAFF receptor TNFRSF13C (BAFF-R) in patients with common variable immunodeficiency. J. Clin. Immunol. 2005, 25, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Hume, A.J.; Abo, K.M.; Werder, R.B.; Villacorta-Martin, C.; Alysandratos, K.D.; Beermann, M.L.; Simone-Roach, C.; Lindstrom-Vautrin, J.; Olejnik, J.; et al. SARS-CoV-2 Infection of Pluripotent Stem Cell-Derived Human Lung Alveolar Type 2 Cells Elicits a Rapid Epithelial-Intrinsic Inflammatory Response. Cell Stem Cell 2020, 27, 962–973. [Google Scholar] [CrossRef]

{kind=link}
| Severe Cases | Non-Severe Cases | Asymptomatic Cases | ||||||
|---|---|---|---|---|---|---|---|---|
| n = 39 | % | n = 82 | % | * p | n = 379 | % | ^ p | |
| Age | ||||||||
| years, mean (standard deviation) | 62.5 (13.3) | 62.2 (15.5) | 0.90 | 60.9 (10.7) | 0.4 | |||
| Gender—no. (%) | ||||||||
| Male | 22 | 56.4 | 52 | 63.4 | 180 | 47.4 | ||
| Female | 17 | 43.6 | 30 | 36.6 | 0.46 | 199 | 52.5 | 0.29 |
| Previous coexisting disease—no. (%) | ||||||||
| 0–2 | 25 | 64.1 | 48 | 58.5 | na | |||
| ≥3 | 7 | 17.9 | 19 | 23.2 | 0.47 | na | ||
| Unknown | 7 | 17.9 | 15 | 18.3 |
| Gene Symbol | Approved Name HGNC | Location | Phenotype | Phenotype MIM Number | Inheritance |
|---|---|---|---|---|---|
| CD19 | CD19 molecule | 16p11.2 | Immunodeficiency, common variable, 3 | 613493 | AR |
| CD81 | CD81 molecule | 11p15.5 | Immunodeficiency, common variable, 6 | 613496 | AR |
| CR2 | Complement C3d receptor 2 | 1q32.2 | Immunodeficiency, common variable, 7 | 614699 | AR |
| ICOS | Inducible T cell costimulator | 2q33.2 | Immunodeficiency, common variable, 1 | 607594 | AR |
| MS4A1 | Membrane spanning 4-domains A1 | 11q12.2 | ?Immunodeficiency, common variable, 5 | 613495 | AR |
| NFKB1 | Nuclear factor-kappa B subunit 1 | 4q24 | Immunodeficiency, common variable, 12 | 616576 | AD |
| NFKB2 | Nuclear factor-kappa B subunit 2 | 10q24.32 | Immunodeficiency, common variable, 10 | 615577 | AD |
| PRKCD | Protein kinase C delta | 3p21.1 | Autoimmune lymphoproliferative syndrome, type III | 615559 | AR |
| TNFRSF13B | TNF receptor superfamily member 13B | 17p11.2 | Immunodeficiency, common variable, 2 | 240500 | AD, AR |
| TNFRSF13C | TNF receptor superfamily member 13C | 22q13.2 | Immunodeficiency, common variable, 4 | 613494 | AR |
| Subgroup | Patient ID | Gene Symbol | HGVS cDNA-Level Nomenclature | HGVS Protein-Level | RefSeq ID | AF Gnomad § | Genotype | CADD Score | InterVar |
|---|---|---|---|---|---|---|---|---|---|
| Severe patients | R_41 | TNFRSF13C | c.475C > T | p.H159Y | rs61756766 | 0.0075 | 0/1 | 27.6 | VUS |
| R_42 | TNFRSF13B | c.542C > A | p.A181E | rs72553883 | 0.0064 | 0/1 | 22.8 | Benign | |
| R_48 | TNFRSF13C | c.475C > T | p.H159Y | rs61756766 | 0.0075 | 0/1 | 27.6 | VUS | |
| R_69 | TNFRSF13C | c.475C > T | p.H159Y | rs61756766 | 0.0075 | 0/1 | 27.6 | VUS | |
| R_158 | TNFRSF13C | c.475C > T | p.H159Y | rs61756766 | 0.0075 | 0/1 | 27.6 | VUS | |
| R_167 | TNFRSF13C | c.475C > T | p.H159Y | rs61756766 | 0.0075 | 0/1 | 27.6 | VUS | |
| R_168 | CR2 | c.1676G > A | p.G559E | rs143614333 | 0.0008 | 0/1 | 24.8 | VUS | |
| R_176 | CR2 | c.1021C > T | p.R341C | rs529311780 | 0.0001 | 0/1 | 28.5 | VUS | |
| Non-severe patients | R_15 | TNFRSF13B | c.310T > C | p.C104R | rs34557412 | 0.0055 | 0/1 | 25.9 | Benign |
| R_46 | CR2 | c.1676G > A | p.G559E | rs143614333 | 0.0008 | 0/1 | 24.8 | VUS | |
| R_99 | CR2 | c.1723C > T | p.R575C | rs758797015 | 0.0000 | 0/1 | 32.0 | VUS | |
| R_127 | TNFRSF13B | c.492C > G | p.Y164 * | rs72553882 | 0.0001 | 0/1 | 37.0 | Pathogenic | |
| R_140 | CD19 | c.5C > T | p.P2L | rs766808956 | 0.0000 | 0/1 | 26.9 | VUS | |
| R_143 | TNFRSF13C | c.475C > T | p.H159Y | rs61756766 | 0.0075 | 0/1 | 27.6 | VUS |
| Severe Cases | Non-Severe Cases | Asymptomatic Cases | Population | |
|---|---|---|---|---|
| N = 38 | N = 82 | N = 375 | N = 283 | |
| Genotype—no. (%) | ||||
| GG | 33 (86.8) | 81 (98.8) | 361 (96.3) | 269 (95.0) |
| GA | 5 (13.2) | 1 (1.2) | 14 (3.7) | 14 (5.0) |
| AA | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 |
| ^ Ptrend | 0.005 | |||
| ^ Common OR | 12.3 | |||
| § Ptrend | 0.008 | |||
| § Common OR | 3.9 | |||
| # Ptrend | 0.04 | |||
| # Common OR | 2.9 | |||
| Allele—no. (%) | ||||
| G | 71 (93.4) | 163 (99.4) | 736 (98.1) | 552 (97.5) |
| A | 5 (6.6) | 1 (0.6) | 14 (1.9) | 14 (2.5) |
| * Pallele | 0.01 | |||
| * OR (95% CI) | 11.5 (1.3–100) | |||
| ° Pallele | 0.02 | |||
| ° OR (95% CI) | 3.7 (1.3–10.6) | |||
| $ Pallele | 0.06 | |||
| $ OR (95% CI) | 2.8 (0.97–7.9) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russo, R.; Andolfo, I.; Lasorsa, V.A.; Cantalupo, S.; Marra, R.; Frisso, G.; Abete, P.; Cassese, G.M.; Servillo, G.; Esposito, G.; et al. The TNFRSF13C H159Y Variant Is Associated with Severe COVID-19: A Retrospective Study of 500 Patients from Southern Italy. Genes 2021, 12, 881. https://doi.org/10.3390/genes12060881
Russo R, Andolfo I, Lasorsa VA, Cantalupo S, Marra R, Frisso G, Abete P, Cassese GM, Servillo G, Esposito G, et al. The TNFRSF13C H159Y Variant Is Associated with Severe COVID-19: A Retrospective Study of 500 Patients from Southern Italy. Genes. 2021; 12(6):881. https://doi.org/10.3390/genes12060881
Chicago/Turabian StyleRusso, Roberta, Immacolata Andolfo, Vito Alessandro Lasorsa, Sueva Cantalupo, Roberta Marra, Giulia Frisso, Pasquale Abete, Gian Marco Cassese, Giuseppe Servillo, Gabriella Esposito, and et al. 2021. "The TNFRSF13C H159Y Variant Is Associated with Severe COVID-19: A Retrospective Study of 500 Patients from Southern Italy" Genes 12, no. 6: 881. https://doi.org/10.3390/genes12060881
APA StyleRusso, R., Andolfo, I., Lasorsa, V. A., Cantalupo, S., Marra, R., Frisso, G., Abete, P., Cassese, G. M., Servillo, G., Esposito, G., Gentile, I., Piscopo, C., Della Monica, M., Fiorentino, G., Russo, G., Cerino, P., Buonerba, C., Pierri, B., Zollo, M., ... Capasso, M. (2021). The TNFRSF13C H159Y Variant Is Associated with Severe COVID-19: A Retrospective Study of 500 Patients from Southern Italy. Genes, 12(6), 881. https://doi.org/10.3390/genes12060881