
Clinical and Genetic Characteristics of Korean Congenital Stationary Night Blindness Patients



Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Clinical Data Collection
2.2. Genetic Analyses
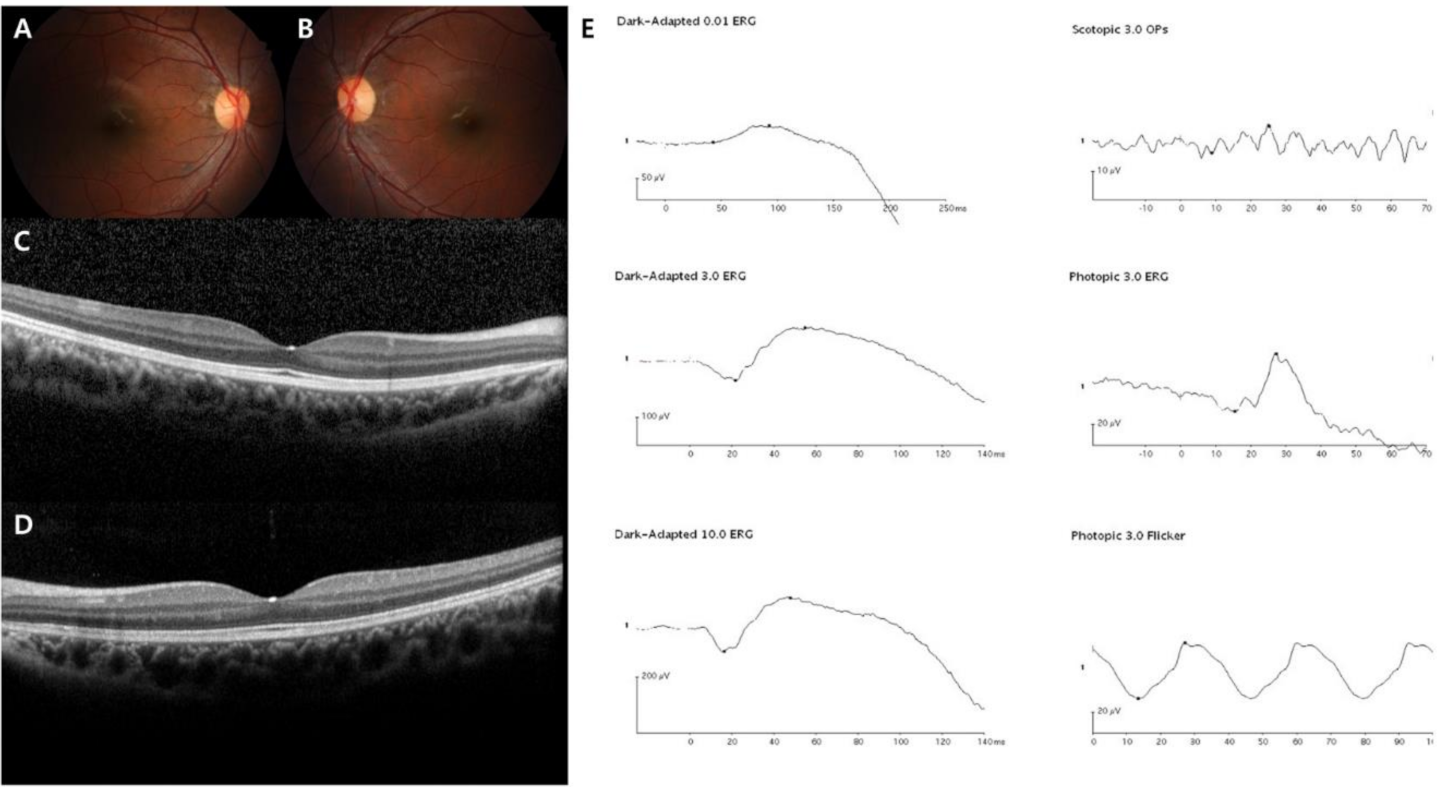
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zeitz, C.; Forster, U.; Neidhardt, J.; Feil, S.; Kalin, S.; Leifert, D.; Flor, P.J.; Berger, W. Night blindness-associated mutations in the ligand-binding, cysteine-rich, and intracellular domains of the metabotropic glutamate receptor 6 abolish protein trafficking. Hum. Mutat. 2007, 28, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, C.; Labs, S.; Lorenz, B.; Forster, U.; Uksti, J.; Kroes, H.Y.; De Baere, E.; Leroy, B.P.; Cremers, F.P.; Wittmer, M.; et al. Genotyping microarray for CSNB-associated genes. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5919–5926. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, C.; Robson, A.G.; Audo, I. Congenital stationary night blindness: An analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog. Retin. Eye Res. 2015, 45, 58–110. [Google Scholar] [CrossRef] [PubMed]
- Miraldi Utz, V.; Pfeifer, W.; Longmuir, S.Q.; Olson, R.J.; Wang, K.; Drack, A.V. Presentation of TRPM1-Associated Congenital Stationary Night Blindness in Children. JAMA Ophthalmol 2018, 136, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Riggs, L.A. Electroretinography in cases of night blindness. Am. J. Ophthalmol. 1954, 38, 70–78. [Google Scholar] [CrossRef]
- Schubert, G.; Bornschein, H. Analysis of the human electroretinogram. Ophthalmologica 1952, 123, 396–413. [Google Scholar] [CrossRef]
- Bijveld, M.M.; Florijn, R.J.; Bergen, A.A.; van den Born, L.I.; Kamermans, M.; Prick, L.; Riemslag, F.C.; van Schooneveld, M.J.; Kappers, A.M.; van Genderen, M.M. Genotype and phenotype of 101 dutch patients with congenital stationary night blindness. Ophthalmology 2013, 120, 2072–2081. [Google Scholar] [CrossRef]
- Dryja, T.P.; Berson, E.L.; Rao, V.R.; Oprian, D.D. Heterozygous missense mutation in the rhodopsin gene as a cause of congenital stationary night blindness. Nat. Genet. 1993, 4, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Szabo, V.; Kreienkamp, H.J.; Rosenberg, T.; Gal, A.P. Gln200Glu, a putative constitutively active mutant of rod α-transducin (GNAT1) in autosomal dominant congenital stationary night blindness. Hum. Mutat. 2007, 28, 741–742. [Google Scholar] [CrossRef]
- Naeem, M.A.; Chavali, V.R.; Ali, S.; Iqbal, M.; Riazuddin, S.; Khan, S.N.; Husnain, T.; Sieving, P.A.; Ayyagari, R.; Riazuddin, S.; et al. GNAT1 associated with autosomal recessive congenital stationary night blindness. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1353–1361. [Google Scholar] [CrossRef]
- Gal, A.; Orth, U.; Baehr, W.; Schwinger, E.; Rosenberg, T. Heterozygous missense mutation in the rod cGMP phosphodiesterase β-subunit gene in autosomal dominant stationary night blindness. Nat. Genet. 1994, 7, 551. [Google Scholar] [CrossRef] [PubMed]
- Manes, G.; Cheguru, P.; Majumder, A.; Bocquet, B.; Senechal, A.; Artemyev, N.O.; Hamel, C.P.; Brabet, P. A truncated form of rod photoreceptor PDE6 β-subunit causes autosomal dominant congenital stationary night blindness by interfering with the inhibitory activity of the γ-subunit. PLoS ONE 2014, 9, e95768. [Google Scholar] [CrossRef]
- Rao, V.R.; Cohen, G.B.; Oprian, D.D. Rhodopsin mutation G90D and a molecular mechanism for congenital night blindness. Nature 1994, 367, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Sieving, P.A.; Richards, J.E.; Naarendorp, F.; Bingham, E.L.; Scott, K.; Alpern, M. Dark-light: Model for nightblindness from the human rhodopsin Gly-90-->Asp mutation. Proc. Natl. Acad. Sci. USA 1995, 92, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Al-Jandal, N.; Farrar, G.J.; Kiang, A.S.; Humphries, M.M.; Bannon, N.; Findlay, J.B.; Humphries, P.; Kenna, P.F. A novel mutation within the rhodopsin gene (Thr-94-Ile) causing autosomal dominant congenital stationary night blindness. Hum. Mutat. 1999, 13, 75–81. [Google Scholar] [CrossRef]
- Zeitz, C.; Gross, A.K.; Leifert, D.; Kloeckener-Gruissem, B.; McAlear, S.D.; Lemke, J.; Neidhardt, J.; Berger, W. Identification and functional characterization of a novel rhodopsin mutation associated with autosomal dominant CSNB. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4105–4114. [Google Scholar] [CrossRef]
- Riazuddin, S.A.; Shahzadi, A.; Zeitz, C.; Ahmed, Z.M.; Ayyagari, R.; Chavali, V.R.; Ponferrada, V.G.; Audo, I.; Michiels, C.; Lancelot, M.E.; et al. A mutation in SLC24A1 implicated in autosomal-recessive congenital stationary night blindness. Am. J. Hum. Genet. 2010, 87, 523–531. [Google Scholar] [CrossRef]
- Bech-Hansen, N.T.; Naylor, M.J.; Maybaum, T.A.; Sparkes, R.L.; Koop, B.; Birch, D.G.; Bergen, A.A.; Prinsen, C.F.; Polomeno, R.C.; Gal, A.; et al. Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat. Genet. 2000, 26, 319–323. [Google Scholar] [CrossRef]
- Pusch, C.M.; Zeitz, C.; Brandau, O.; Pesch, K.; Achatz, H.; Feil, S.; Scharfe, C.; Maurer, J.; Jacobi, F.K.; Pinckers, A.; et al. The complete form of X-linked congenital stationary night blindness is caused by mutations in a gene encoding a leucine-rich repeat protein. Nat. Genet. 2000, 26, 324–327. [Google Scholar] [CrossRef]
- Ivanova, M.E.; Zolnikova, I.V.; Gorgisheli, K.V.; Atarshchikov, D.S.; Ghosh, P.; Barh, D. Novel frameshift mutation in NYX gene in a Russian family with complete congenital stationary night blindness. Ophthalmic. Genet. 2019, 40, 558–563. [Google Scholar] [CrossRef]
- Dryja, T.P.; McGee, T.L.; Berson, E.L.; Fishman, G.A.; Sandberg, M.A.; Alexander, K.R.; Derlacki, D.J.; Rajagopalan, A.S. Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proc. Natl. Acad. Sci. USA 2005, 102, 4884–4889. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, C.; van Genderen, M.; Neidhardt, J.; Luhmann, U.F.; Hoeben, F.; Forster, U.; Wycisk, K.; Matyas, G.; Hoyng, C.B.; Riemslag, F.; et al. Mutations in GRM6 cause autosomal recessive congenital stationary night blindness with a distinctive scotopic 15-Hz flicker electroretinogram. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4328–4335. [Google Scholar] [CrossRef] [PubMed]
- Audo, I.; Kohl, S.; Leroy, B.P.; Munier, F.L.; Guillonneau, X.; Mohand-Said, S.; Bujakowska, K.; Nandrot, E.F.; Lorenz, B.; Preising, M.; et al. TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 2009, 85, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Sergouniotis, P.I.; Michaelides, M.; Mackay, D.S.; Wright, G.A.; Devery, S.; Moore, A.T.; Holder, G.E.; Robson, A.G.; Webster, A.R. Recessive mutations of the gene TRPM1 abrogate ON bipolar cell function and cause complete congenital stationary night blindness in humans. Am. J. Hum. Genet. 2009, 85, 711–719. [Google Scholar] [CrossRef]
- Van Genderen, M.M.; Bijveld, M.M.; Claassen, Y.B.; Florijn, R.J.; Pearring, J.N.; Meire, F.M.; McCall, M.A.; Riemslag, F.C.; Gregg, R.G.; Bergen, A.A.; et al. Mutations in TRPM1 are a common cause of complete congenital stationary night blindness. Am. J. Hum. Genet. 2009, 85, 730–736. [Google Scholar] [CrossRef]
- Al-Hujaili, H.; Taskintuna, I.; Neuhaus, C.; Bergmann, C.; Schatz, P. Long-term follow-up of retinal function and structure in TRPM1-associated complete congenital stationary night blindness. Mol. Vis. 2019, 25, 851–858. [Google Scholar] [PubMed]
- AlTalbishi, A.; Zelinger, L.; Zeitz, C.; Hendler, K.; Namburi, P.; Audo, I.; Sheffer, R.; Yahalom, C.; Khateb, S.; Banin, E.; et al. TRPM1 Mutations are the Most Common Cause of Autosomal Recessive Congenital Stationary Night Blindness (CSNB) in the Palestinian and Israeli Populations. Sci. Rep. 2019, 9, 12047. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, Y.; Zeevi, D.A.; Lam, B.L.; Scher, S.Y.; Bringer, R.; Cherki, B.; Cohen, C.C.; Muallem, H.; Chiang, J.P.; Pantrangi, M.; et al. A founder deletion in the TRPM1 gene associated with congenital stationary night blindness and myopia is highly prevalent in Ashkenazi Jews. Hum. Genome Var. 2019, 6, 45. [Google Scholar] [CrossRef]
- Hayashi, T.; Mizobuchi, K.; Kikuchi, S.; Nakano, T. Novel biallelic TRPM1 variants in an elderly patient with complete congenital stationary night blindness. Doc. Ophthalmol. 2020. [Google Scholar] [CrossRef]
- Lee, Y.J.; Joo, K.; Seong, M.W.; Park, K.H.; Park, S.S.; Woo, S.J. Congenital Stationary Night Blindness due to Novel TRPM1 Gene Mutations in a Korean Patient. Korean J. Ophthalmol. 2020, 34, 170–172. [Google Scholar] [CrossRef]
- Audo, I.; Bujakowska, K.; Orhan, E.; Poloschek, C.M.; Defoort-Dhellemmes, S.; Drumare, I.; Kohl, S.; Luu, T.D.; Lecompte, O.; Zrenner, E.; et al. Whole-exome sequencing identifies mutations in GPR179 leading to autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 2012, 90, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Peachey, N.S.; Ray, T.A.; Florijn, R.; Rowe, L.B.; Sjoerdsma, T.; Contreras-Alcantara, S.; Baba, K.; Tosini, G.; Pozdeyev, N.; Iuvone, P.M.; et al. GPR179 is required for depolarizing bipolar cell function and is mutated in autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 2012, 90, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, C.; Jacobson, S.G.; Hamel, C.P.; Bujakowska, K.; Neuille, M.; Orhan, E.; Zanlonghi, X.; Lancelot, M.E.; Michiels, C.; Schwartz, S.B.; et al. Whole-exome sequencing identifies LRIT3 mutations as a cause of autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 2013, 92, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Bech-Hansen, N.T.; Naylor, M.J.; Maybaum, T.A.; Pearce, W.G.; Koop, B.; Fishman, G.A.; Mets, M.; Musarella, M.A.; Boycott, K.M. Loss-of-function mutations in a calcium-channel alpha1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat. Genet. 1998, 19, 264–267. [Google Scholar] [CrossRef]
- Strom, T.M.; Nyakatura, G.; Apfelstedt-Sylla, E.; Hellebrand, H.; Lorenz, B.; Weber, B.H.; Wutz, K.; Gutwillinger, N.; Ruther, K.; Drescher, B.; et al. An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat. Genet. 1998, 19, 260–263. [Google Scholar] [CrossRef]
- Boycott, K.M.; Pearce, W.G.; Bech-Hansen, N.T. Clinical variability among patients with incomplete X-linked congenital stationary night blindness and a founder mutation in CACNA1F. Can. J. Ophthalmol. 2000, 35, 204–213. [Google Scholar] [CrossRef]
- Nakamura, M.; Ito, S.; Terasaki, H.; Miyake, Y. Novel CACNA1F mutations in Japanese patients with incomplete congenital stationary night blindness. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1610–1616. [Google Scholar]
- Mahmood, U.; Mejecase, C.; Ali, S.M.A.; Moosajee, M.; Kozak, I. A Novel Splice-Site Variant in CACNA1F Causes a Phenotype Synonymous with Aland Island Eye Disease and Incomplete Congenital Stationary Night Blindness. Genes 2021, 12, 17. [Google Scholar] [CrossRef]
- Marmor, M.F.; Fulton, A.B.; Holder, G.E.; Miyake, Y.; Brigell, M.; Bach, M. International Society for Clinical Electrophysiology of, V. ISCEV Standard for full-field clinical electroretinography (2008 update). Doc. Ophthalmol. 2009, 118, 69–77. [Google Scholar] [CrossRef]
- Seong, M.W.; Seo, S.H.; Yu, Y.S.; Hwang, J.M.; Cho, S.I.; Ra, E.K.; Park, H.; Lee, S.J.; Kim, J.Y.; Park, S.S. Diagnostic application of an extensive gene panel for Leber congenital amaurosis with severe genetic heterogeneity. J. Mol. Diagn. 2015, 17, 100–105. [Google Scholar] [CrossRef]
- Kim, M.S.; Joo, K.; Seong, M.W.; Kim, M.J.; Park, K.H.; Park, S.S.; Woo, S.J. Genetic Mutation Profiles in Korean Patients with Inherited Retinal Diseases. J. Korean. Med. Sci. 2019, 34, e161. [Google Scholar] [CrossRef]
- Plagnol, V.; Curtis, J.; Epstein, M.; Mok, K.Y.; Stebbings, E.; Grigoriadou, S.; Wood, N.W.; Hambleton, S.; Burns, S.O.; Thrasher, A.J.; et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 2012, 28, 2747–2754. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef]
- ClinVar. NM_022567.2(NYX):c.38-1_38delinsTT. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/496942/ (accessed on 13 March 2021).
- Hove, M.N.; Kilic-Biyik, K.Z.; Trotter, A.; Gronskov, K.; Sander, B.; Larsen, M.; Carroll, J.; Bech-Hansen, T.; Rosenberg, T. Clinical Characteristics, Mutation Spectrum, and Prevalence of Aland Eye Disease/Incomplete Congenital Stationary Night Blindness in Denmark. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6861–6869. [Google Scholar] [CrossRef] [PubMed]
- Rim, J.H.; Lee, S.T.; Gee, H.Y.; Lee, B.J.; Choi, J.R.; Park, H.W.; Han, S.H.; Han, J. Accuracy of Next-Generation Sequencing for Molecular Diagnosis in Patients With Infantile Nystagmus Syndrome. JAMA Ophthalmol. 2017, 135, 1376–1385. [Google Scholar] [CrossRef] [PubMed]
- Zito, I.; Allen, L.E.; Patel, R.J.; Meindl, A.; Bradshaw, K.; Yates, J.R.; Bird, A.C.; Erskine, L.; Cheetham, M.E.; Webster, A.R.; et al. Mutations in the cacna1f and nyx genes in british csnbx families. Hum. Mutat. 2003, 21, 169. [Google Scholar] [CrossRef]
- Wutz, K.; Sauer, C.; Zrenner, E.; Lorenz, B.; Alitalo, T.; Broghammer, M.; Hergersberg, M.; de la Chapelle, A.; Weber, B.H.; Wissinger, B.; et al. Thirty distinct CACNA1F mutations in 33 families with incomplete type of XLCSNB and Cacna1f expression profiling in mouse retina. Eur. J. Hum. Genet. 2002, 10, 449–456. [Google Scholar] [CrossRef]
- Wang, X.; Zein, W.M.; D’Souza, L.; Roberson, C.; Wetherby, K.; He, H.; Villarta, A.; Turriff, A.; Johnson, K.R.; Fann, Y.C. Applying next generation sequencing with microdroplet pcr to determine the disease-causing mutations in retinal dystrophies. BMC Ophthalmol. 2017, 17, 157. [Google Scholar] [CrossRef]
- Sun, W.; Huang, L.; Xu, Y.; Xiao, X.; Li, S.; Jia, X.; Gao, B.; Wang, P.; Guo, X.; Zhang, Q. Exome Sequencing on 298 Probands With Early-Onset High Myopia: Approximately One-Fourth Show Potential Pathogenic Mutations in RetNet Genes. Investig. Ophthalmol. Vis. Sci. 2015, 56, 8365–8372. [Google Scholar] [CrossRef] [PubMed]
- Almutairi, F.; Almeshari, N.; Ahmad, K.; Magliyah, M.S.; Schatz, P. Congenital stationary night blindness: An update and review of the disease spectrum in Saudi Arabia. Acta Ophthalmol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Koike, C.; Obara, T.; Uriu, Y.; Numata, T.; Sanuki, R.; Miyata, K.; Koyasu, T.; Ueno, S.; Funabiki, K.; Tani, A.; et al. TRPM1 is a component of the retinal ON bipolar cell transduction channel in the mGluR6 cascade. Proc. Natl. Acad. Sci. USA 2010, 107, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Orhan, E.; Prezeau, L.; El Shamieh, S.; Bujakowska, K.M.; Michiels, C.; Zagar, Y.; Vol, C.; Bhattacharya, S.S.; Sahel, J.A.; Sennlaub, F.; et al. Further insights into GPR179: Expression, localization, and associated pathogenic mechanisms leading to complete congenital stationary night blindness. Investig. Ophthalmol. Vis. Sci. 2013, 54, 8041–8050. [Google Scholar] [CrossRef]
- Specht, D.; Wu, S.B.; Turner, P.; Dearden, P.; Koentgen, F.; Wolfrum, U.; Maw, M.; Brandstatter, J.H.; tom Dieck, S. Effects of presynaptic mutations on a postsynaptic Cacna1s calcium channel colocalized with mGluR6 at mouse photoreceptor ribbon synapses. Investig. Ophthalmol. Vis. Sci. 2009, 50, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kerov, V.; Haeseleer, F.; Majumder, A.; Artemyev, N.; Baker, S.A.; Lee, A. Dysregulation of Ca(v)1.4 channels disrupts the maturation of photoreceptor synaptic ribbons in congenital stationary night blindness type 2. Channels (Austin) 2013, 7, 514–523. [Google Scholar] [CrossRef]
- Muradov, K.G.; Granovsky, A.E.; Artemyev, N.O. Mutation in rod PDE6 linked to congenital stationary night blindness impairs the enzyme inhibition by its γ-subunit. Biochemistry 2003, 42, 3305–3310. [Google Scholar] [CrossRef]
- Marmor, M.F.; Zeitz, C. Riggs-type dominant congenital stationary night blindness: ERG findings, a new GNAT1 mutation and a systemic association. Doc. Ophthalmol. 2018, 137, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, C.; Méjécase, C.; Stévenard, M.; Michiels, C.; Audo, I.; Marmor, M.F. A Novel Heterozygous Missense Mutation in GNAT1 Leads to Autosomal Dominant Riggs Type of Congenital Stationary Night Blindness. BioMed Res. Int. 2018, 2018. [Google Scholar] [CrossRef]
- Ba-Abbad, R.; Holder, G.E.; Robson, A.G.; Neveu, M.M.; Waseem, N.; Arno, G.; Webster, A.R. Isolated rod dysfunction associated with a novel genotype of CNGB1. Am. J. Ophthalmol. Case Rep. 2019, 14, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Yagasaki, K.; Horiguchi, M.; Kawase, Y.; Kanda, T. Congenital stationary night blindness with negative electroretinogram. A new classification. Arch. Ophthalmol. 1986, 104, 1013–1020. [Google Scholar] [CrossRef]
- Allen, L.E.; Zito, I.; Bradshaw, K.; Patel, R.J.; Bird, A.C.; Fitzke, F.; Yates, J.R.; Trump, D.; Hardcastle, A.J.; Moore, A.T. Genotype-phenotype correlation in British families with X linked congenital stationary night blindness. Br. J. Ophthalmol. 2003, 87, 1413–1420. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Sex | Age at Exam (y) | Age at Symptom Onset (y) | Age at Last Visit (y) | Diagnosis | Gene | Inheritance | SE (D) | Initial BCVA (logMAR) | Final BCVA (logMAR) | Strabismus | Nystagmus | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OD | OS | OD | OS | OD | OS | ||||||||||
| 1 | M | 21 | 1 | 22 | Riggs | CNGB1 | AR | −1.0 | 1.5 | 0.1 | 0.1 | 0.1 | 0.1 | No | No |
| 2 | M | 9 | 8 | 12 | Riggs | GNAT1 | AD | 0.5 | 0.5 | 0.2 | 0.2 | 0.2 | 0.2 | No | No |
| 3 † | M | 19 | 4 | 28 | Complete | TRPM1 | AR | −7.5 | −6.5 | 0.4 | 0.4 | 0.3 | 0.3 | No | No |
| 4 | M | 3 | 1 | 7 | Complete | NYX | XL | −6.5 | −5.5 | poor | poor | poor | poor | No | Yes |
| 5 | M | 5 | 4 | 8 | Complete | NYX | XL | −9.5 | −9.5 | 0.7 | 0.7 | 0.6 | 0.6 | Yes | No |
| 6 | M | 3 | 1 | 7 | Incomplete | CACNA1F | XL | 3.0 | 3.0 | 0.7 | 0.8 | 0.7 | 0.7 | No | Yes |
| 7 | M | 3 | 1 | 8 | Incomplete | CACNA1F | XL | −1.0 | 0.5 | 1.0 | 0.8 | 0.9 | 0.8 | Yes | Yes |
| 8 | M | 3 | 1 | 6 | Incomplete | CACNA1F | XL | 0.5 | 1.0 | 0.8 | 0.7 | 0.7 | 0.7 | Yes | Yes |
| 9 | M | 3 | 2 | 5 | Incomplete | CACNA1F | XL | −1.0 | −1.5 | 0.9 | 0.9 | 0.8 | 0.8 | No | Yes |
| 10 | M | 3 | 2 | 13 | Incomplete | CACNA1F | XL | −10.5 | −9.5 | 0.7 | 0.6 | 0.7 | 0.6 | Yes | No |
| 11 † | M | 4 | 1 | 13 | Incomplete | CACNA1F | XL | −11.5 | −10.0 | 0.6 | 0.7 | 0.6 | 0.6 | No | Yes |
| 12 | M | 4 | 2 | 12 | Incomplete | CACNA1F | XL | −3.0 | −3.0 | 0.6 | 0.6 | 0.6 | 0.6 | Yes | No |
| 13 | M | 5 | 4 | 9 | Incomplete | CACNA1F | XL | −1.5 | −1.5 | 0.6 | 0.6 | 0.5 | 0.5 | No | Yes |
| 14 | M | 5 | 1 | 8 | Incomplete | CACNA1F | XL | −2.0 | −1.5 | 1.0 | 1.0 | 0.9 | 0.9 | Yes | Yes |
| 15 ‡ | M | 7 | 1 | 22 | Incomplete | CACNA1F | XL | 2.5 | 0.5 | 0.7 | 0.7 | 0.7 | 0.7 | No | Yes |
| 16 | M | 7 | 4 | 18 | Incomplete | CACNA1F | XL | −9.5 | −8.5 | 0.7 | 0.6 | 0.6 | 0.6 | No | Yes |
| 17 | M | 9 | 6 | 11 | Incomplete | CACNA1F | XL | 1.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | No | No |
| 18 | M | 10 | 8 | 11 | Incomplete | CACNA1F | XL | −1.5 | −0.5 | 1.2 | 1.5 | 1.0 | 1.5 | No | No |
| 19 | M | 13 | 6 | 22 | Incomplete | CACNA1F | XL | −0.5 | −0.5 | 0.6 | 0.6 | 0.6 | 0.6 | No | Yes |
| Case | Nystagmus | Case | Nystagmus | ||||||||||||
| 3 | 3–4 Hz pendular-LBJ bilateral symmetric | 13 | gaze evoked nystagmus | ||||||||||||
| 6 | Gaze evoked nystagmus | 14 | 3 Hz UBJ bilateral symmetric | ||||||||||||
| 7 | 1–2 Hz RBJ-LBJ intermittent bilateral symmetric | 15 | 4 Hz multiplanar nystagmus bilateral symmetric | ||||||||||||
| 8 | 5 Hz small amplitude pendular nystagmus | 16 | gaze evoked nystagmus | ||||||||||||
| 9 | 4 Hz fine amplitude pendular nystagmus | 19 | gaze evoked nystagmus | ||||||||||||
| 11 | 3–4 Hz LBJ bilateral symmetric | ||||||||||||||
| Type | Gene | Age at Symptom Onset (y) | Sex (M:F) | SE | Initial BCVA (logMAR) | Final BCVA (logMAR) | Strabismus (Yes:No) | Nystagmus (Yes:No) | |
|---|---|---|---|---|---|---|---|---|---|
| Riggs (n = 2) | CNGB1 GNAT1 | 4.5 ± 3.5 | 2:0 | 0.3 ± 0.9 | 0.15 ±0.05 | 0.15 ± 0.05 | 0:2 | 0:2 | |
| Complete (n = 1) | TRPM1 | 4 | 1:0 | −7.0 | 0.4 | 0.3 | 0:1 | 0:1 | |
| Complete (n = 2) | NYX | 2.5 ± 1.5 | 2:0 | −7.8 ± 1.8 | 0.70 ± 0.00 | 0.60 ± 0.00 | 1:1 (50%) | 1:1 (50%) | |
| Incomplete (n = 14) | CACNA1F | 2.7 ± 2.2 | 12:0 | −2.6 ± 4.4 | 0.79 ± 0.21 | 0.74 ± 0.19 | 5:9 (36%) | 10:4 (71%) | |
| Type | Dark-Adapted 0.01 ERG | Dark-Adapted 3.0 ERG | Light-Adapted 3.0 ERG | 30-Hz Flicker ERG | |||||
| A-wave amplitude (μV) | B-wave amplitude (μV) | A-wave amplitude (μV) | B-wave amplitude (μV) | B/A ratio | A-wave amplitude (μV) | B-wave amplitude (μV) | Amplitude (μV) | Double peak (Yes:No) | |
| Riggs (n = 2) | 0.9 ± 0.2 | 37.2 ± 3.5 | 66.9 ± 8.3 | 54.1 ± 4.7 | 0.81 ± 0.05 | 19.5 ± 2.1 | 55.9 ± 3.7 | 51.8 ± 4.9 | 0:2 (0%) |
| Complete (n = 1) | 0 | 0 | 184.1 ± 38.3 | 86.3 ± 1.2 | 0.49 ± 0.09 | 46.8 ± 4.1 | 85.6 ± 0.3 | 74.5 ± 5.1 | 0:1 (0%) |
| Complete (n = 2) | 0 | 0 | 31.8 ± 6.2 | 18.9 ± 1.1 | 0.61 ± 0.08 | 6.6 ± 0.7 | 12.7 ± 3.8 | 11.7 ± 2.6 | 0:2 (0%) |
| Incomplete (n = 14) | 3.2 ± 0.7 | 25.4 ± 2.8 | 43.7 ± 6.0 | 25.3 ± 3.3 | 0.58 ± 0.02 | 3.8 ± 0.5 | 6.5 ± 0.3 | 5.7 ± 0.9 | 9:5 (64%) |
| Age-matched normal controls (n = 30) | 32.97±11.35 | 234.74 ± 98.65 | 150.02 ± 61.05 | 359.77 ± 89.67 | 2.68 ± 0.99 | 21.56 ± 9.67 | 70.40 ± 21.97 | 74.44 ± 21.83 | 0:30 (0%) |
| No. | Gene | Transcript | Nucleotide Change | Amino Acid Change | Zygosity | Segregation | CADD | FATHMM | SpliceAI | MAF (Gnomad) | Domain | Novel Variant |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | CNGB1 | NM_001297.5 | c.2544delG | p.(Leu849Cysfs*15) | Hetero | - | - | 0.987 | - | 2/249580 | - | Novel |
| c.1035-1G>A | - | Hetero | - | 32 | 0.547 | 0.96 | Not found | - | Novel | |||
| 2 | GNAT1 | NM_000172.4 | c.753C>A | p.(Asn251Lys) | Hetero | - | 24.5 | 0.894 | - | 13/251156 | α subunit of G proteins | Novel |
| 3 | TRPM1 | NM_002420.6 | c.3280C>T | p.(Arg1094*) | Hetero | - | 38 | 0.937 | - | 1/249530 | - | Lee et al. [30] |
| c.3794delA | p.(Asn1265Ilefs*42) | 26.8 | 0.991 | - | Not found | - | ||||||
| 4 | NYX | NM_022567.2 | c.182_183insT | p.(Cys62Valfs*53) | Hemi | Maternal | 32 | 0.981 | - | Not found | Leucine-rich repeat | Novel |
| 5 | NYX | NM_022567.2 | c.38-1_ 38delGCinsTT | Hemi | Maternal | 14.16 | - | - | Not found | - | ClinVar [45] | |
| 6 | CACNA1F | NM_005183.2 | exon(13–23) deletion | Hemi | - | - | - | - | Not found | - | Novel | |
| 7 | CACNA1F | NM_005183.2 | c.1301C>T | p.(Ala434Val) | Hemi | Maternal | 17.47 | 0.143 | 0.80 | Not found | - | Hove et al. [46] |
| 8 | CACNA1F | NM_005183.2 | c.2175_2179delins27 | p.(Gly726llefs*61) | Hemi | Maternal | 27.3 | 0.999 | - | Not found | Ion transport | Novel |
| 9 | CACNA1F | NM_005183.2 | c.1910+1G>A | Hemi | - | 24.9 | 0.976 | 0.95 | Not found | Ion transport | Novel | |
| 10 | CACNA1F | NM_005183.2 | c.4049G>A | p.(Gly1350Asp) | Hemi | - | 27.2 | 0.987 | - | Not found | Voltage-dependent calcium channel | Kim et al. [41] |
| 11 | CACNA1F | NM_005183.2 | c.342delC | p.(Phe115Serfs*22) | Hemi | Maternal | 25.6 | 0.940 | - | Not found | - | Rim et al. [47] |
| 12 | CACNA1F | NM_005183.2 | c.2914C>T | p.(Arg972*) | Hemi | - | 12.6 | 0.352 | - | Not found | - | Zito et al. [48] |
| 13 | CACNA1F | NM_005183.2 | c.4042-1G>T | Hemi | Maternal | 34 | 0.993 | 0.98 | Not found | Voltage-dependent calcium channel | Novel | |
| 14 | CACNA1F | NM_005183.2 | c.1910+1G>A | Hemi | - | 24.9 | 0.976 | 0.95 | Not found | Ion transport | Novel | |
| 15 | CACNA1F | NM_005183.2 | c.5479C>T | p.(Arg1827*) | Hemi | Maternal | 36 | 0.202 | - | 1/183192 | - | Wutz et al. [49] |
| 16 | CACNA1F | NM_005183.2 | c.2576+1G>A | Hemi | - | 34 | 0.993 | 0.88 | 1/62946 | - | Wang et al. [50] | |
| 17 | CACNA1F | NM_005183.2 | c.926G>A | p.(Gly309Asp) | Hemi | - | 26.2 | 0.974 | - | Not found | Ion transport | Sun et al. [51] |
| 18 | CACNA1F | NM_005183.2 | c.2761C>A | p.(Leu921IIe) | Hemi | Maternal | 25.3 | 0.976 | - | Not found | Ion transport | Novel |
| 19 | CACNA1F | NM_005183.2 | c.2767-1G>C | Hemi | - | 35 | 0.994 | 0.99 | Not found | Ion transport | Novel |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.-M.; Joo, K.; Han, J.; Woo, S.-J. Clinical and Genetic Characteristics of Korean Congenital Stationary Night Blindness Patients. Genes 2021, 12, 789. https://doi.org/10.3390/genes12060789
Kim H-M, Joo K, Han J, Woo S-J. Clinical and Genetic Characteristics of Korean Congenital Stationary Night Blindness Patients. Genes. 2021; 12(6):789. https://doi.org/10.3390/genes12060789
Chicago/Turabian StyleKim, Hyeong-Min, Kwangsic Joo, Jinu Han, and Se-Joon Woo. 2021. "Clinical and Genetic Characteristics of Korean Congenital Stationary Night Blindness Patients" Genes 12, no. 6: 789. https://doi.org/10.3390/genes12060789
APA StyleKim, H.-M., Joo, K., Han, J., & Woo, S.-J. (2021). Clinical and Genetic Characteristics of Korean Congenital Stationary Night Blindness Patients. Genes, 12(6), 789. https://doi.org/10.3390/genes12060789