
Expanding the Phenotypic and Genotypic Spectrum of Bietti Crystalline Dystrophy

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
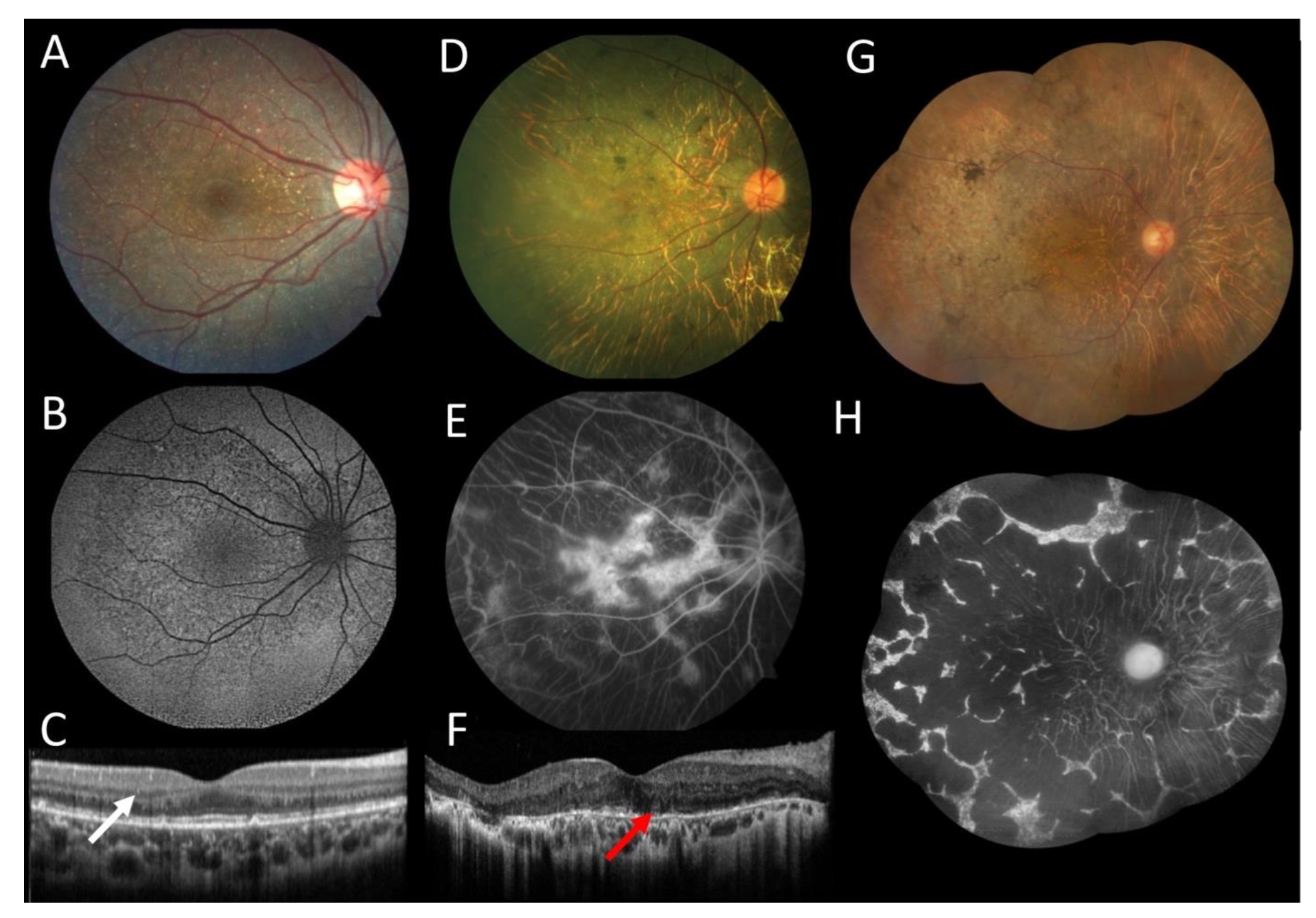
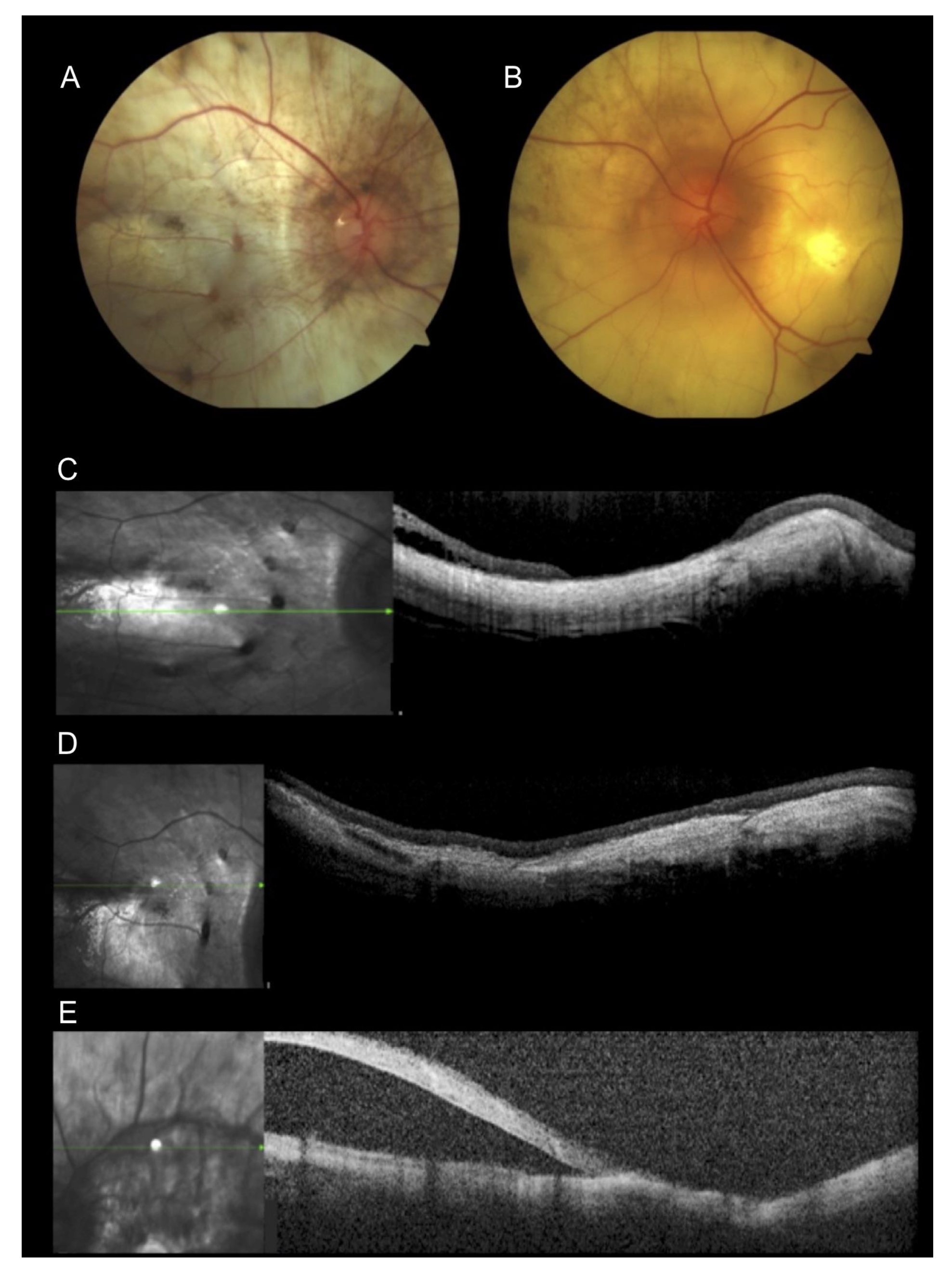
3. Results
3.1. Case 1
3.2. Case 2
3.3. Case 3
3.4. Case 4
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Garcia-Garcia, G.P.; Martinez-Rubio, M.; Moya-Moya, M.A.; Perez-Santonja, J.J.; Escribano, J. Identification of novel CYP4V2 genotypes associated with Bietti crystalline dystrophy and atypical anterior segment phenotypes in Spanish patients. Acta Ophthalmol. 2018, 96, e865–e873. [Google Scholar] [CrossRef]
- Lockhart, C.M.; Smith, T.B.; Yang, P.; Naidu, M.; Rettie, A.E.; Nath, A.; Weleber, R.; Kelly, E.J. Longitudinal characterization of function and structure of Bietti crystalline dystrophy: Report on a novel homozygous mutation in CYP4V2. Br. J. Ophthalmol. 2018, 102, 187–194. [Google Scholar] [CrossRef]
- Li, A.; Jiao, X.; Munier, F.L.; Schorderet, D.F.; Yao, W.; Iwata, F.; Hayakawa, M.; Kanai, A.; Chen, M.S.; Lewis, R.A.; et al. Bietti crystalline dystrophy is caused by mutations in the novel gene CYP4V2. Am. J. Hum. Genet. 2004, 74, 817–826. [Google Scholar] [CrossRef]
- Ng, D.S.C.; Lai, T.Y.Y.; Ng, T.K.; Pang, C.P. Genetics of Bietti crystalline dystrophy. Asia Pac. J. Ophthalmol. 2016, 5, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.J.; Weleber, R.G.; Klein, M.L.; Welch, R.B.; Green, W.R. Bietti’s crystalline dystrophy. A clinicopathologic correlative study. Arch. Ophthalmol. 1989, 107, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Kaiser-Kupfer, M.I.; Chan, C.C.; Markello, T.C.; Crawford, M.A.; Caruso, R.C.; Csaky, K.G.; Guo, J.; Gahl, W.A. Clinical biochemical and pathologic correlations in Bietti’s crystalline dystrophy. Am. J. Ophthalmol. 1994, 118, 569–582. [Google Scholar] [CrossRef]
- Oishi, A.; Oishi, M.; Miyata, M.; Hirashima, T.; Hasegawa, T.; Numa, S.; Tsujikawa, A. Multimodal imaging for differential diagnosis of Bietti crystalline dystrophy. Ophthalmol. Retin. 2018, 2, 1071–1077. [Google Scholar] [CrossRef]
- Savige, J.; Sheth, S.; Leys, A.; Nicholson, A.; Mack, H.G.; Colville, D. Ocular features in Alport syndrome: Pathogenesis and clinical significance. Clin. J. Am. Soc. Nephrol. 2015, 10, 703–709. [Google Scholar] [CrossRef]
- Yuzawa, M.; Mae, Y.; Matsui, M. Bietti’s crystalline retinopathy. Ophthalmic Paediatr. Genet. 1986, 7, 9–20. [Google Scholar] [CrossRef]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant Interpret and supporting evidence. Nucleic Acids Res. 2018, 4, D1062–D1067. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Lin, J.; Nishiguchi, K.M.; Nakamura, M.; Dryja, T.P.; Berson, E.L.; Miyake, Y. Recessive mutations in the CYP4V2 gene in East Asian and Middle Eastern patients with Bietti crystalline corneoretinal dystrophy. J. Med. Genet. 2005, 42, e38. [Google Scholar] [CrossRef] [PubMed]
- Salzano, F.M.; Sans, M. Interethnic admixture and the evolution of Latin American populations. Genet. Mol. Biol. 2014, 37, 151–170. [Google Scholar] [CrossRef]
- Brazilian Citizens with Japanese Ancestry Is Now at 1.5 Million. Available online: http://www.brazil.gov.br/about-brazil/news/2017/06/brazil-has-1-5-million-citizens-of-japanese-origin (accessed on 7 March 2021).
- Xiao, X.; Mai, G.; Li, S.; Guo, X.; Zhang, Q. Identification of CYP4V2 mutation in 21 families and overview of mutation spectrum in Bietti crystalline corneoretinal dystrophy. Biochem. Biophys. Res. Commun. 2011, 409, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.Y.; Ng, T.K.; Tam, P.O.; Yam, G.H.; Ngai, J.W.; Chan, W.M.; Liu, D.T.L.; Lam, D.S.C.; Pang, C.P. Genotype-phenotype analysis of Bietti’s crystalline dystrophy in patients with CYP4V2 mutations. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5212–5220. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, K.; Dong, B.; Peng, X.; Li, Q.; Jiang, F.; Xie, Y.; Tian, L.; Li, Y. Comprehensive screening of CYP4V2 in a cohort of Chinese patients with Bietti crystalline dystrophy. Mol. Vis. 2018, 24, 700–711. [Google Scholar]
- Jiao, X.; Li, A.; Jin, Z.B.; Wang, X.; Iannaccone, A.; Traboulsi, E.I.; Gorin, M.B.; Simonelli, F.; Hejtmancik, J.F. Identification and population history of CYP4V2 mutations in patients with Bietti crystalline corneoretinal dystrophy. Eur. J. Hum. Genet. 2017, 25, 461–471. [Google Scholar] [CrossRef]
- Astuti, G.D.N.; Sun, V.; Bauwens, M.; Zobor, D.; Leroy, B.P.; Omar, A.; Jurklies, B.; Lopez, I.; Ren, H.; Yazar, V.; et al. Novel insights into the molecular pathogenesis of CYP4V2-associated Bietti’s retinal dystrophy. Mol. Genet. Genom. Med. 2015, 3, 14–29. [Google Scholar] [CrossRef]
- Rossi, S.; Testa, F.; Li, A.; Yaylacioğlu, F.; Gesualdo, C.; Hejtmancik, J.F.; Simonelli, F. Clinical and genetic features in Italian Bietti crystalline dystrophy patients. Br. J. Ophthalmol. 2013, 97, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.C.; Koh, A.H.C.; Aung, T.; Yong, V.H.K.; Yeung, K.; Ang, C.-L.; Vithana, E.N. Characterization of Bietti crystalline dystrophy patients with CYP4V2 mutations. Investig. Opthalmol. Vis. Sci. 2005, 46, 3812–3816. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Cooper, G.M.; Goode, D.L.; Ng, S.B.; Sidow, A.; Bamshad, M.J.; Shendure, J.; Nickerson, D.A. Single-nucleotide evolutionary constraint scores highlight disease-causing mutations. Nat. Methods 2010, 7, 250–251. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Yokoi, Y.; Sato, K.; Aoyagi, H.; Takahashi, Y.; Yamagami, M.; Nakazawa, M. A novel compound heterozygous mutation in the CYP4V2 gene in a Japanese patient with Bietti’s crystalline corneoretinal dystrophy. Case Rep. Ophthalmol. 2011, 2, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Halford, S.; Liew, G.; Mackay, D.S.; Sergouniotis, P.I.; Holt, R.; Broadgate, S.; Volpi, E.V.; Ocaka, L.; Robson, A.G.; Holder, G.E.; et al. Detailed phenotypic and genotypic characterization of Bietti crystalline dystrophy. Ophthalmology 2014, 121, 1174–1184. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Patient | Age, Sex | c.DNA Change in CYP4V2 | Protein Change | BCVA OD OS | Crystalline Deposits | OCT Findings |
|---|---|---|---|---|---|---|
| 1 | 19y, female | c.802-8_810delinsGC homozygous | p.? | 20/20 20/20 | Retina | Intraretinal hyperreflective crystals |
| 2 | 54y, female | c.518 T > G c.802-8_806del | p.Leu173Trp p.? | 20/50 20/400 | Retina | Outer retinal atrophy, few intraretinal crystals, and tubulations |
| 3 | 69y, female | c.518T > G homozygous | p.Leu174Trp | 20/150 20/150 | Retina | Not available |
| 4 | 59y, male | c.1169G > T homozygous | p.Arg390Leu | LP HM | None | Extensive atrophy and diffuse thinning. Central retinal detachment |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Palma, M.M.; Motta, F.L.; Salles, M.V.; Texeira, C.H.M.; Gomes, A.V.; Casaroli-Marano, R.; Sallum, J.M.F. Expanding the Phenotypic and Genotypic Spectrum of Bietti Crystalline Dystrophy. Genes 2021, 12, 713. https://doi.org/10.3390/genes12050713
da Palma MM, Motta FL, Salles MV, Texeira CHM, Gomes AV, Casaroli-Marano R, Sallum JMF. Expanding the Phenotypic and Genotypic Spectrum of Bietti Crystalline Dystrophy. Genes. 2021; 12(5):713. https://doi.org/10.3390/genes12050713
Chicago/Turabian Styleda Palma, Mariana Matioli, Fabiana Louise Motta, Mariana Vallim Salles, Caio Henrique Marques Texeira, André V. Gomes, Ricardo Casaroli-Marano, and Juliana Maria Ferraz Sallum. 2021. "Expanding the Phenotypic and Genotypic Spectrum of Bietti Crystalline Dystrophy" Genes 12, no. 5: 713. https://doi.org/10.3390/genes12050713
APA Styleda Palma, M. M., Motta, F. L., Salles, M. V., Texeira, C. H. M., Gomes, A. V., Casaroli-Marano, R., & Sallum, J. M. F. (2021). Expanding the Phenotypic and Genotypic Spectrum of Bietti Crystalline Dystrophy. Genes, 12(5), 713. https://doi.org/10.3390/genes12050713