
Meta-Analysis of Gene Popularity: Less Than Half of Gene Citations Stem from Gene Regulatory Networks



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Pubmed Data Retrieval and Pre-Processing
2.2. Citation Distribution Analysis
2.3. Gene Family Analysis
2.4. Gene Homology Analysis
2.5. Gene Expression Analysis
2.6. Gene Co-Expression Network Analysis
2.7. Protein–Protein Interaction (PPI) Network Analysis
2.8. Gene Essentiality Analysis
2.9. Gene Chromatin Proximity Analysis
2.10. GWAS Analysis
2.11. COSMIC Analysis
2.12. The Total Model (Linear)
2.13. The Total Model (Nonlinear)
2.14. Creation of Online Data Visualizer
2.15. Drug Availability Analysis
3. Results
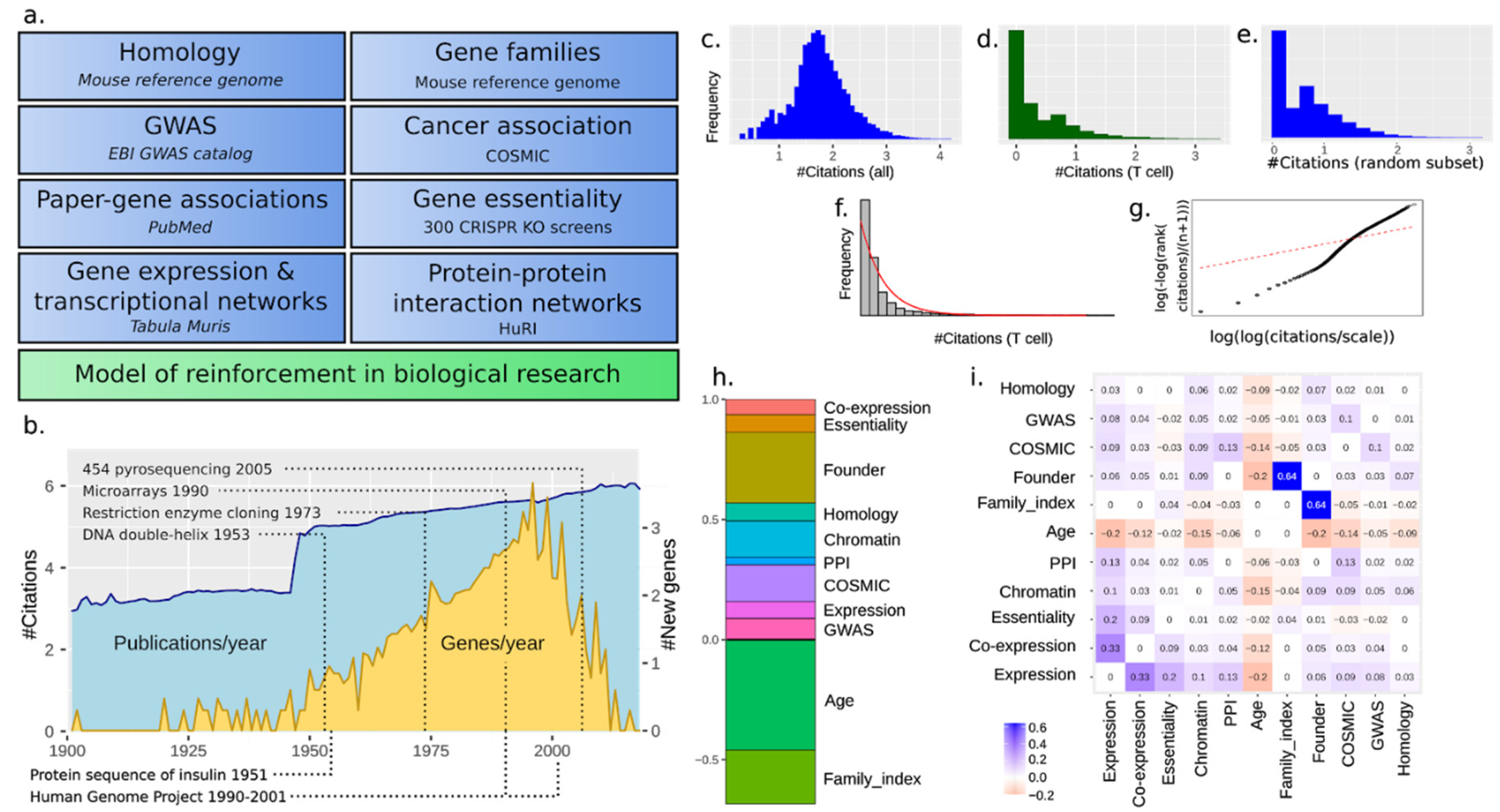
3.1. The Scholastic Gene Coverage Follows Matthew Principle under Cellular Context
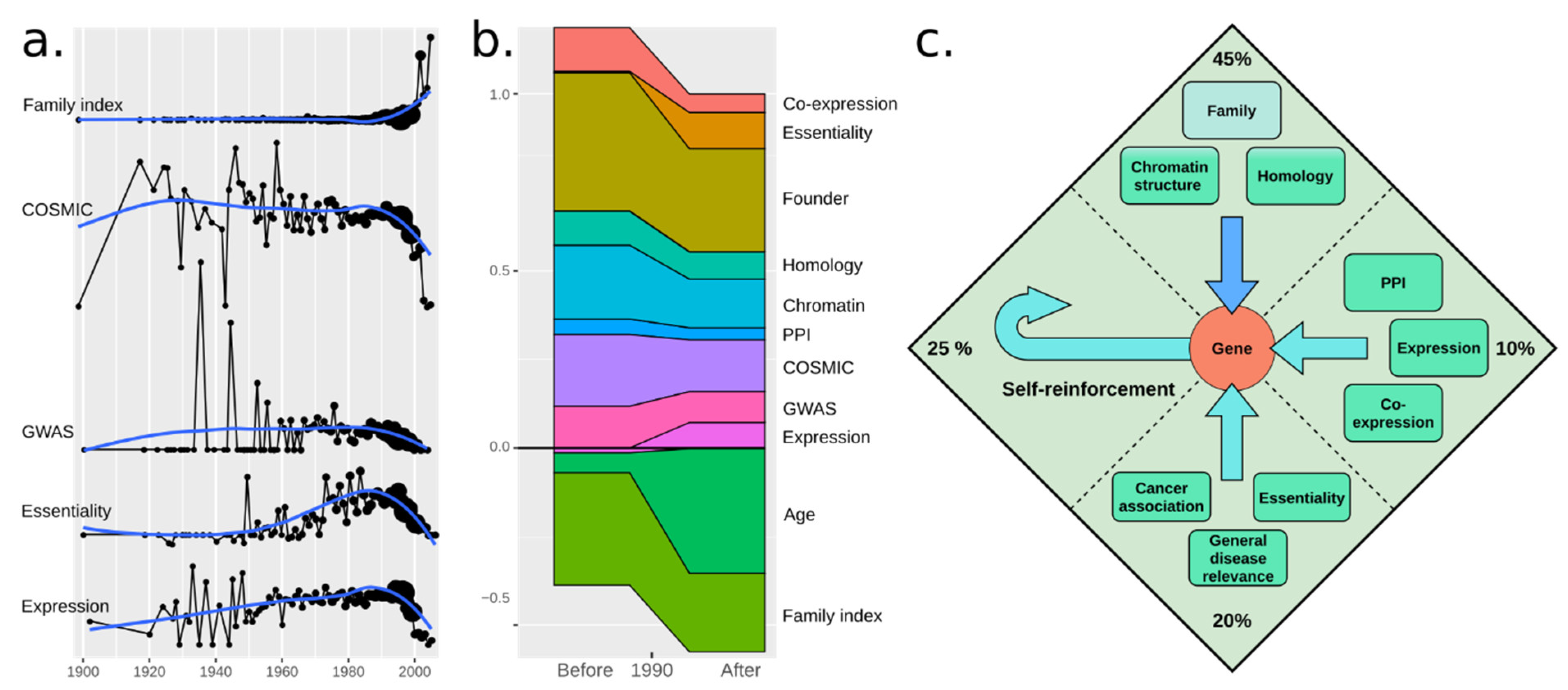
3.2. A Total Model Integrates Sources of Reinforcement
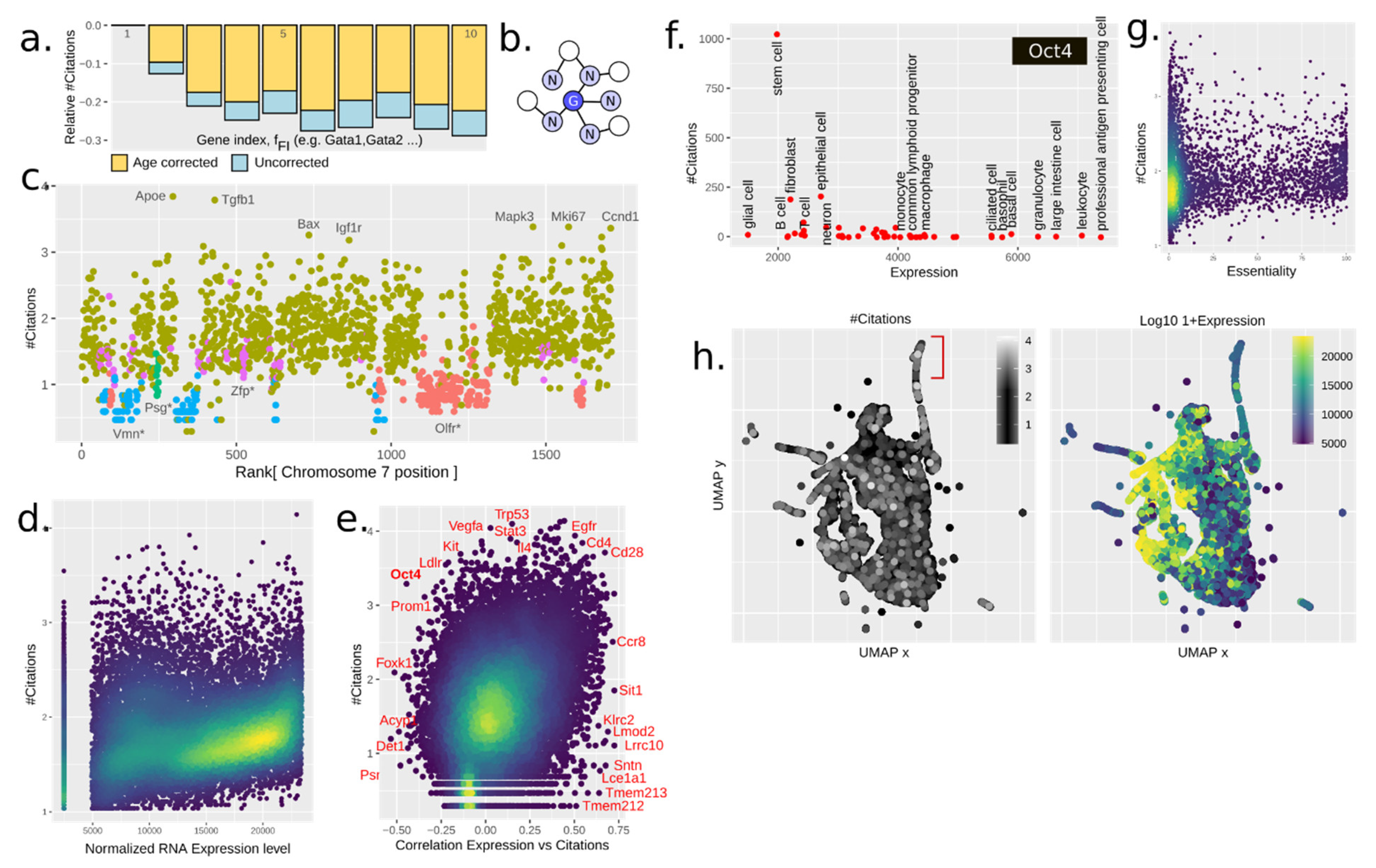
3.3. Paralogy and Gene Family Linkage Are Strong Drivers of Gene Popularity
3.4. The Chromatin Structure Influences Citations in Several Manners
3.5. Gene Expression Drives Citations
3.6. Disease Association and Essentiality Make Up 20% of All Citations
3.7. Gene Regulatory Networks (Grns) Are Weak Reinforcers
3.8. Social Reinforcement Has Increased over Time
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stoeger, T.; Gerlach, M.; Morimoto, R.I.; Amaral, L.A.N. Large-scale investigation of the reasons why potentially important genes are ignored. PLoS Biol. 2018, 16, e2006643. [Google Scholar] [CrossRef]
- Mingers, J.; Leydesdorff, L. A review of theory and practice in scientometrics. Eur. J. Oper. Res. 2015, 246, 1–19. [Google Scholar] [CrossRef]
- Bailón-Moreno, R.; Jurado-Alameda, E.; Ruiz-Baños, R.; Courtial, J.P. Bibliometric laws: Empirical flaws of fit. Scientometrics 2005, 63, 209–229. [Google Scholar] [CrossRef]
- Kim, S.; Yeganova, L.; Wilbur, W.J. Meshable: Searching PubMed abstracts by utilizing MeSH and MeSH-derived topical terms. Bioinformatics 2016, 32, 3044–3046. [Google Scholar] [CrossRef] [PubMed]
- Venables, W.N.; Ripley, B.D. Modern Applied Statistics with S. Statistics and Computing, 4th ed.; Springer: Berlin, Germany, 2002. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Favera, R.D.; Califano, A. ARACNE: An Algorithm for the Reconstruction of Gene Regulatory Networks in a Mammalian Cellular Context. BMC Bioinform. 2006, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- The Tabula Muris Consortium; Overall Coordination; Logistical Coordination; Organ Collection and Processing; Library Preparation and Sequencing; Computational Data Analysis; Cell Type Annotation; Writing Group; Supplemental Text Writing Group; Principal Investigators; et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018, 562, 367–372. [Google Scholar] [CrossRef]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.-A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2019, 37, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Luck, K.; Kim, D.-K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B.; et al. A reference map of the human binary protein interactome. Nature 2020, 580, 402–408. [Google Scholar] [CrossRef]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nat. Cell Biol. 2019, 568, 511–516. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. PyTorch: An Imperative Style, High-Performance Deep Learning Library. In Advances in Neural Information Processing Systems 32; Wallach, H., Larochelle, H., Beygelzimer, A., d’Alché-Buc, F., Fox, E., Garnett, R., Eds.; Curran Associates Inc.: Red Hook, NY, USA, 2019; pp. 8026–8037. [Google Scholar]
- Ribeiro, M.T.; Singh, S.; Guestrin, C. “Why Should I Trust You?”: Explaining the Predictions of Any Classifier. In Proceedings of the 2016 Conference of the North American Chapter of the Association for Computational Linguistics: Demonstrations, San Diego, CA, USA, 12–17 June 2016; Association for Computational Linguistics: Stroudsburg, PA, USA, 2016; pp. 97–101. [Google Scholar]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; Association for Computing Machinery: New York, NY, USA, 2016; pp. 785–794. [Google Scholar]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Assempour, N. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Watson, J.D.; Crick, F.H.C. Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid. Nat. Cell Biol. 1953, 171, 737–738. [Google Scholar] [CrossRef]
- Cohen, S.N.; Chang, A.C.Y.; Boyer, H.W.; Helling, R.B. Construction of Biologically Functional Bacterial Plasmids In Vitro. Proc. Natl. Acad. Sci. USA 1973, 70, 3240–3244. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, S.M.; Lee, S.-I. A unified approach to interpreting model predictions. In Proceedings of the Advances in Neural In-formation Processing Systems 30 (NIPS 2017), Long Beach, CA, USA, 4–9 December 2017; Guyon, I., Luxburg, U.V., Bengio, S., Wallach, H., Fergus, R., Vishwanathan, S., Garnett, R., Eds.; NeurIPS: Long Beach, CA, USA, 2017; pp. 4765–4774. [Google Scholar]
- Ribeiro, M.T.; Singh, S.; Guestrin, C. “Why Should I Trust You?”: Explaining the Predictions of Any Classifier. arXiv 2016, arXiv:1602.04938. Available online: https://arxiv.org/abs/1602.04938 (accessed on 9 August 2016).
- Lee, G.R.; E Fields, P.; Griffin, T.J.; A Flavell, R. Regulation of the Th2 Cytokine Locus by a Locus Control Region. Immunity 2003, 19, 145–153. [Google Scholar] [CrossRef]
- Niwa, H.; Miyazaki, J.; Smith, A.G. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat. Genet. 2000, 24, 372–376. [Google Scholar] [CrossRef]
- GBD 2016 Causes of Death Collaborators. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1151–1210. [Google Scholar] [CrossRef]
- Lopez, A.D.; Christopher, C.J. The global burden of disease, 1990–2020. Nat. Med. 1998, 4, 1241–1243. [Google Scholar] [CrossRef]
- Sullivan, W. The Institute for the Study of Non–Model Organisms and other fantasies. Mol. Biol. Cell 2015, 26, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Merton, R.K. The Matthew Effect in Science: The reward and communication systems of science are considered. Science 1968, 159, 56–63. [Google Scholar] [CrossRef]
- Deng, Z.; Wang, H.; Chen, Z.; Wang, T. Bibliometric Analysis of Dendritic Epidermal T Cell (DETC) Research From 1983 to 2019. Front. Immunol. 2020, 11, 259. [Google Scholar] [CrossRef] [PubMed]
- Romero, L.; Portillo-Salido, E. Trends in Sigma-1 Receptor Research: A 25-Year Bibliometric Analysis. Front. Pharmacol. 2019, 10, 564. [Google Scholar] [CrossRef] [PubMed]
- Stoeger, T.; Amaral, L.A.N. COVID-19 research risks ignoring important host genes due to pre-established research patterns. eLife 2020, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nat. Cell Biol. 2005, 437, 376–380. [Google Scholar] [CrossRef]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative Monitoring of Gene Expression Patterns with a Complementary DNA Microarray. Science 1995, 270, 467–470. [Google Scholar] [CrossRef]
- Human Genome Project: Sequencing the Human Genome. Learn Science at Scitable. Available online: https://www.nature.com/scitable/topicpage/dna-sequencing-technologies-key-to-the-human-828/ (accessed on 5 June 2020).
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Mouse Genome Sequencing Consortium; Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef]
- Karlebach, G.; Shamir, R. Modelling and analysis of gene regulatory networks. Nat. Rev. Mol. Cell Biol. 2008, 9, 770–780. [Google Scholar] [CrossRef]
- Paananen, J.; Fortino, V. An omics perspective on drug target discovery platforms. Briefings Bioinform. 2020, 21, 1937–1953. [Google Scholar] [CrossRef]
- Collins, F.S.; Green, E.D.; Guttmacher, A.E.; Guyer, M.S.; US National Human Genome Research Institute. A vision for the future of genomics research. Nature 2003, 422, 835–847. [Google Scholar] [CrossRef]
- Zeng, A.; Shen, Z.; Zhou, J.; Fan, Y.; Di, Z.; Wang, Y.; Stanley, H.E.; Havlin, S. Increasing trend of scientists to switch between topics. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Kaelin, W.G. Common pitfalls in preclinical cancer target validation. Nat. Rev. Cancer 2017, 17, 425–440. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mihai, I.S.; Das, D.; Maršalkaite, G.; Henriksson, J. Meta-Analysis of Gene Popularity: Less Than Half of Gene Citations Stem from Gene Regulatory Networks. Genes 2021, 12, 319. https://doi.org/10.3390/genes12020319
Mihai IS, Das D, Maršalkaite G, Henriksson J. Meta-Analysis of Gene Popularity: Less Than Half of Gene Citations Stem from Gene Regulatory Networks. Genes. 2021; 12(2):319. https://doi.org/10.3390/genes12020319
Chicago/Turabian StyleMihai, Ionut Sebastian, Debojyoti Das, Gabija Maršalkaite, and Johan Henriksson. 2021. "Meta-Analysis of Gene Popularity: Less Than Half of Gene Citations Stem from Gene Regulatory Networks" Genes 12, no. 2: 319. https://doi.org/10.3390/genes12020319
APA StyleMihai, I. S., Das, D., Maršalkaite, G., & Henriksson, J. (2021). Meta-Analysis of Gene Popularity: Less Than Half of Gene Citations Stem from Gene Regulatory Networks. Genes, 12(2), 319. https://doi.org/10.3390/genes12020319