
Detection of Somatic Mutations with ddPCR from Liquid Biopsy of Colorectal Cancer Patients

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Controls Selection
2.2. DNA Isolation from Whole Blood Samples
2.3. DNA Isolation from Tumor Samples
2.4. Cell-Free DNA Isolation from Serum Samples and Fluorometric Quantification
2.5. ddPCR Assay for Quantification of Cell-Free DNA and Leukocyte Contamination Evaluation
2.6. Somatic Mutation Detection (ddPCR) in Tumor and Serum DNA Samples
2.7. Sanger Sequencing
2.8. Statistical Analysis
3. Results
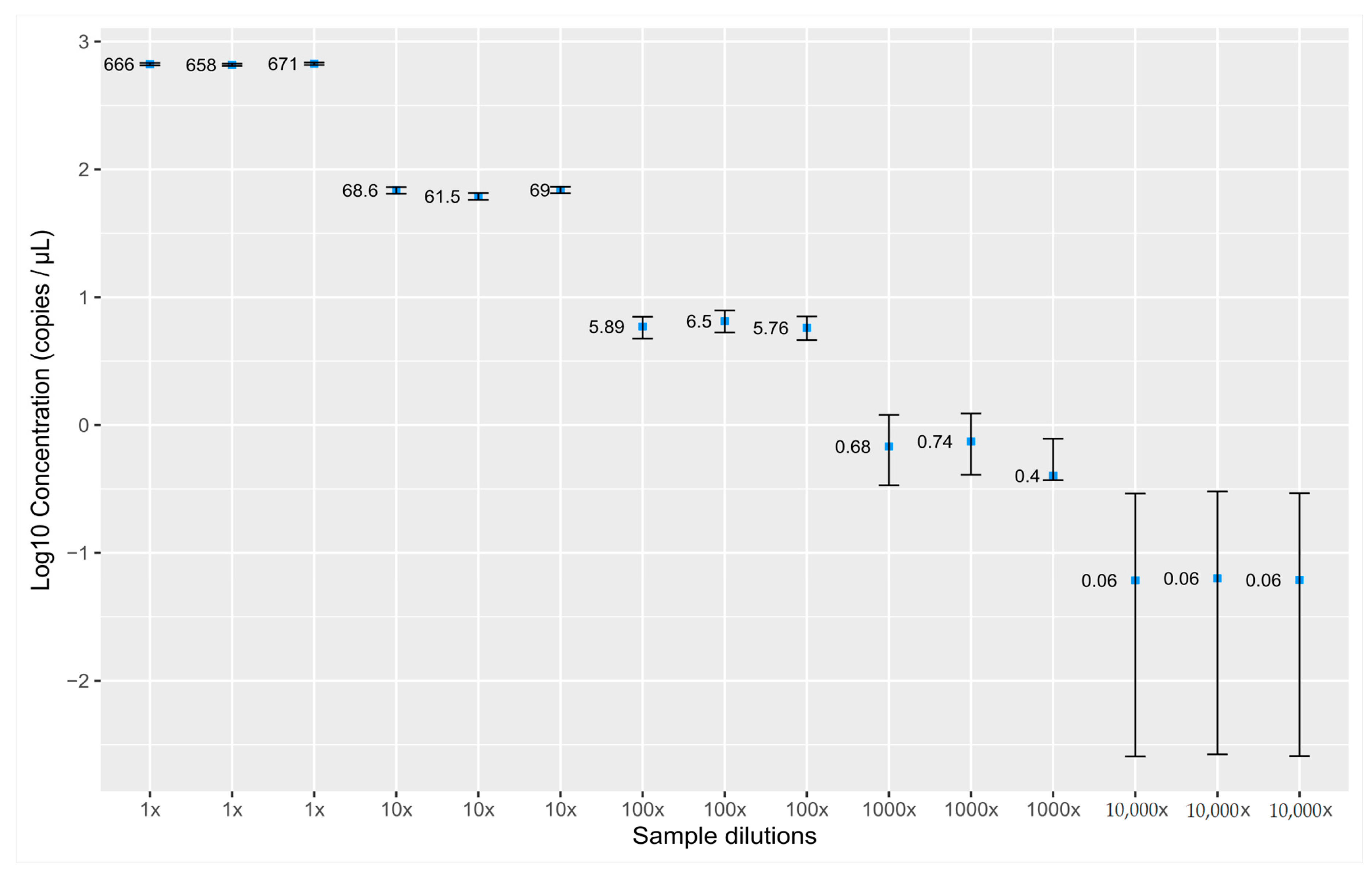
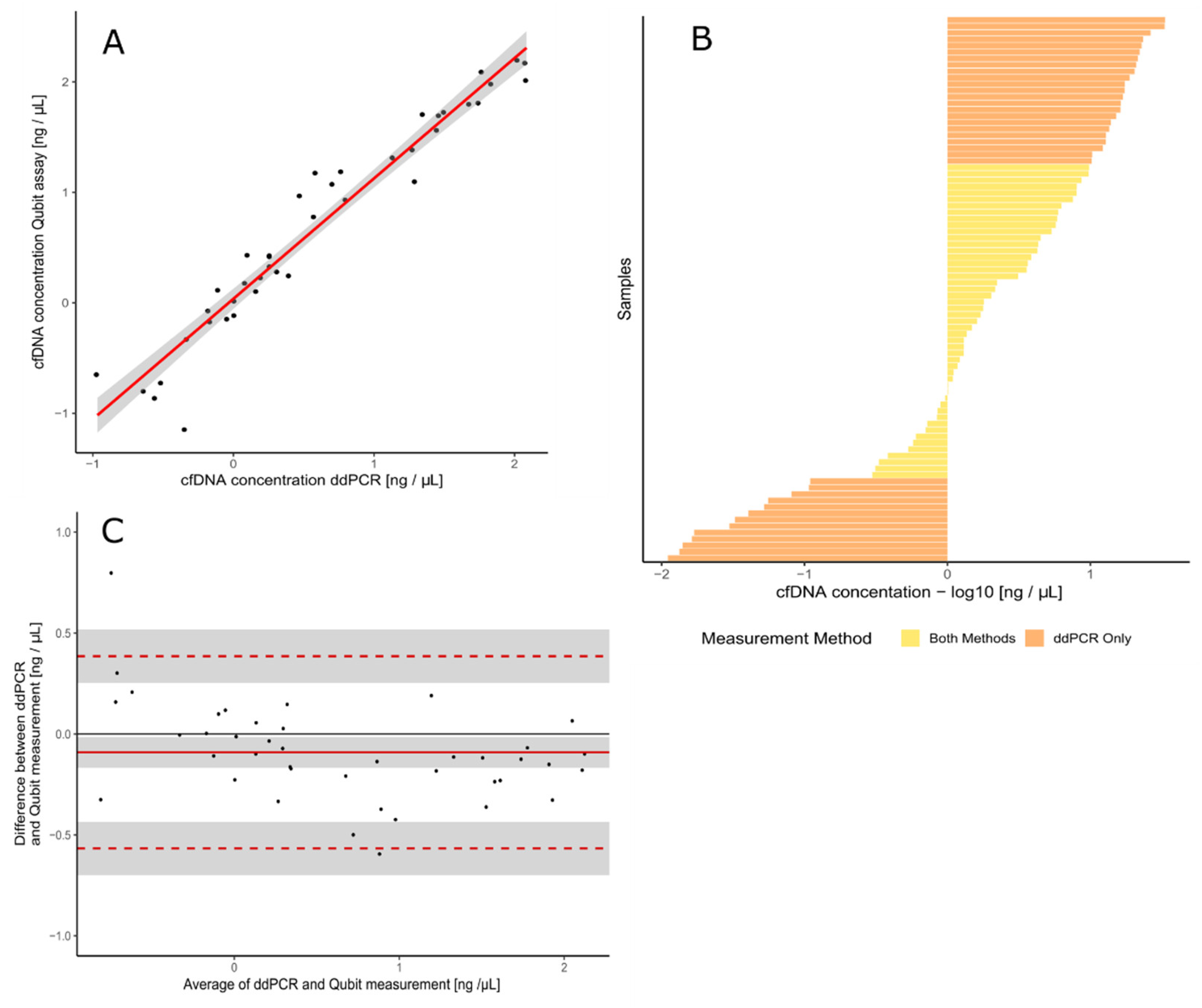
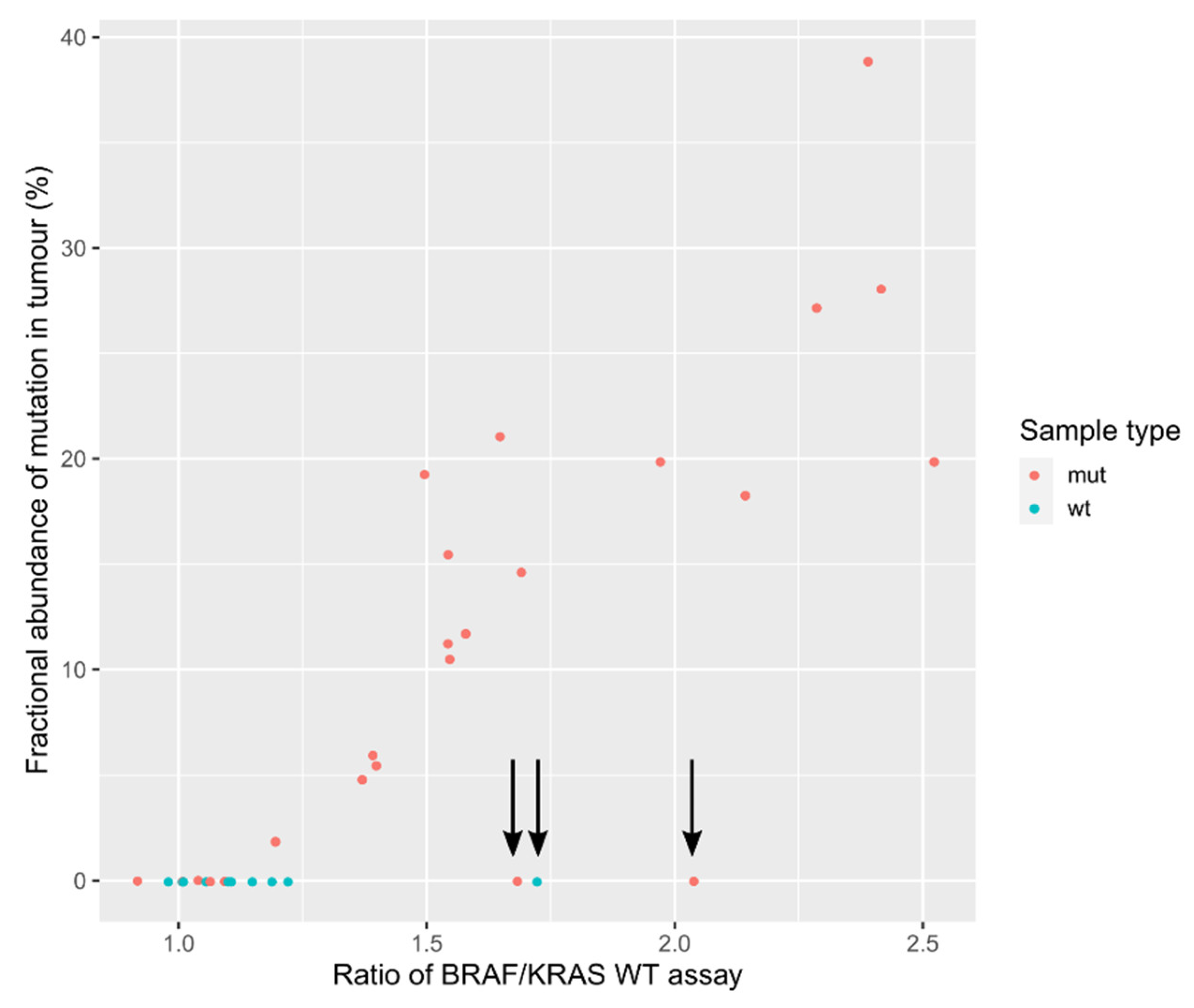
3.1. Validation of the Assay for cfDNA Concentration Measurement
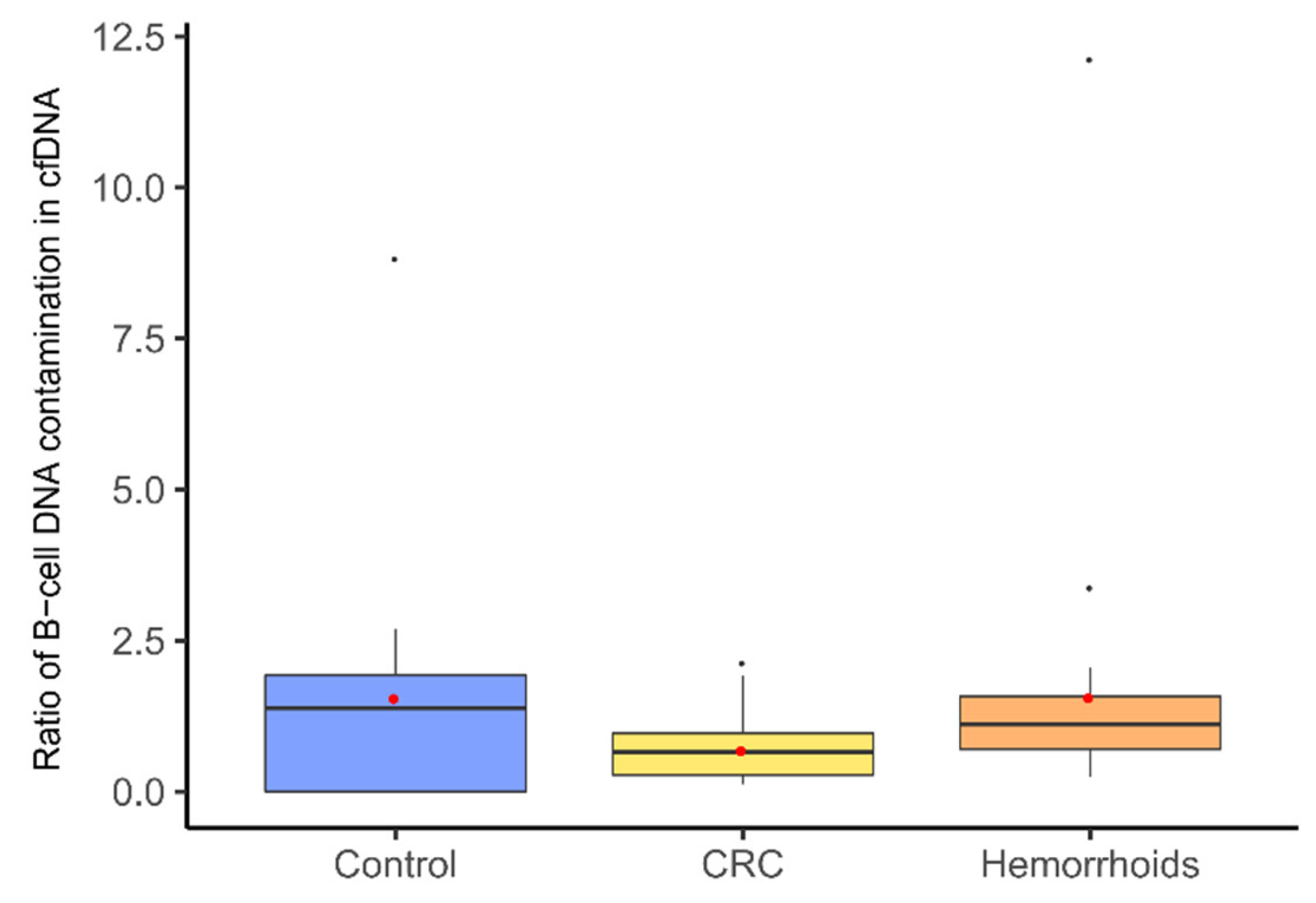
3.2. Measurement of cfDNA Concentration and Quality Assessment with B-Cell Assay
3.3. Patients
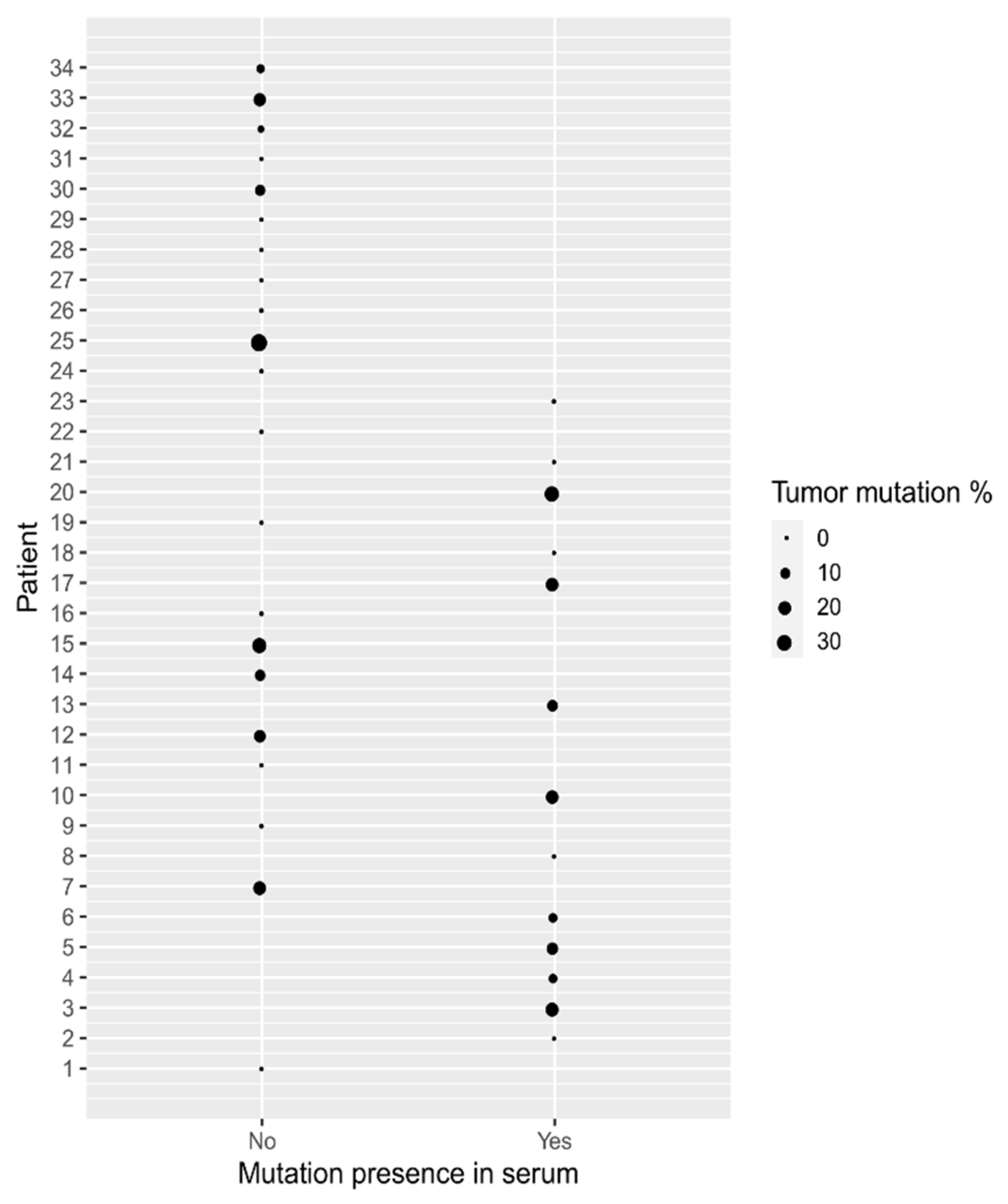
3.4. Primary Tumor Mutation Status
3.5. Matching cfDNA Mutation Status with a Primary Tumor Sample
3.6. Evaluation of Mutation Detection Specificity
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Salomon, D.S.; Brandt, R.; Ciardiello, F.; Normanno, N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit. Rev. Oncol. 1995, 19, 183–232. [Google Scholar] [CrossRef]
- Spano, J.-P.; Lagorce, C.; Atlan, D.; Milano, G.; Domont, J.; Benamouzig, R.; Attar, A.; Benichou, J.; Martin, A.; Morere, J.-F.; et al. Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann. Oncol. 2005, 16, 102–108. [Google Scholar] [CrossRef]
- Bonomi, P.D.; Buckingham, L.; Coon, J. Selecting Patients for Treatment with Epidermal Growth Factor Tyrosine Kinase Inhibitors. Clin. Cancer Res. 2007, 13, 4606s–4612s. [Google Scholar] [CrossRef] [PubMed]
- Amado, R.G.; Wolf, M.; Peeters, M.; Van Cutsem, E.; Siena, S.; Freeman, D.J.; Juan, T.; Sikorski, R.; Suggs, S.; Radinsky, R.; et al. Wild-TypeKRASIs Required for Panitumumab Efficacy in Patients with Metastatic Colorectal Cancer. J. Clin. Oncol. 2008, 26, 1626–1634. [Google Scholar] [CrossRef]
- Lièvre, A.; Bachet, J.-B.; Boige, V.; Cayre, A.; Le Corre, D.; Buc, E.; Ychou, M.; Bouché, O.; Landi, B.; Louvet, C.; et al. KRASMutations As an Independent Prognostic Factor in Patients with Advanced Colorectal Cancer Treated with Cetuximab. J. Clin. Oncol. 2008, 26, 374–379. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aguilar, E.A.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef]
- Cheng, F.; Su, L.; Qian, C. Circulating tumor DNA: A promising biomarker in the liquid biopsy of cancer. Oncotarget 2016, 7, 48832–48841. [Google Scholar] [CrossRef]
- Zhang, B.O.; Xu, C.-W.; Shao, Y.; Wang, H.-T.; Wu, Y.-F.; Song, Y.-Y.; Li, X.-B.; Zhang, Z.; Wang, W.-J.; Li, L.-Q.; et al. Comparison of droplet digital PCR and conventional quantitative PCR for measuring EGFR gene mutation. Exp. Ther. Med. 2015, 9, 1383–1388. [Google Scholar] [CrossRef]
- Beije, N.; Helmijr, J.C.; Weerts, M.J.; Beaufort, C.M.; Wiggin, M.; Marziali, A.; Verhoef, C.; Sleijfer, S.; Jansen, M.P.; Martens, J.W. Somatic mutation detection using various targeted detection assays in paired samples of circulating tumor DNA, primary tumor and metastases from patients undergoing resection of colorectal liver metastases. Mol. Oncol. 2016, 10, 1575–1584. [Google Scholar] [CrossRef]
- Santoni-Rugiu, E.; Melchior, L.C.; Urbanska, E.M.; Jakobsen, J.N.; De Stricker, K.; Grauslund, M.; Sørensen, J.B. Intrinsic resistance to EGFR-Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small Cell Lung Cancer: Differences and Similarities with Acquired Resistance. Cancers 2019, 11, 923. [Google Scholar] [CrossRef]
- Valpione, S.; Gremel, G.; Mundra, P.; Middlehurst, P.; Galvani, E.; Girotti, M.; Lee, R.; Garner, G.; Dhomen, N.; Lorigan, P.; et al. Plasma total cell-free DNA (cfDNA) is a surrogate biomarker for tumour burden and a prognostic biomarker for survival in metastatic melanoma patients. Eur. J. Cancer 2018, 88, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pallisgaard, N.; Spindler, K.-L.G.; Andersen, R.F.; Brandslund, I.; Jakobsen, A. Controls to validate plasma samples for cell free DNA quantification. Clin. Chim. Acta 2015, 446, 141–146. [Google Scholar] [CrossRef]
- Warren, J.D.; Xiong, W.; Bunker, A.M.; Vaughn, C.P.; Furtado, L.V.; Roberts, W.L.; Fang, J.C.; Samowitz, W.S.; Heichman, K.A. Septin 9 methylated DNA is a sensitive and specific blood test for colorectal cancer. BMC Med. 2011, 9, 133. [Google Scholar] [CrossRef]
- LeComte, T.; Berger, A.; Zinzindohoué, F.; Micard, S.; Landi, B.; Blons, H.; Beaune, P.; Cugnenc, P.-H.; Laurent-Puig, P. Detection of free-circulating tumor-associated DNA in plasma of colorectal cancer patients and its association with prognosis. Int. J. Cancer 2002, 100, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.M.; Lefort, F.; McManus, R.; Daly, J.; Keeling, P.W.N.; Weir, D.G.; Kelleher, D. A prospective study of circulating mutant KRAS2 in the serum of patients with colorectal neoplasia: Strong prognostic indicator in postoperative follow up. Gut 2003, 52, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.-L.; Christie, M.; et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 2016, 8, 346ra92. [Google Scholar] [CrossRef]
- Lefebure, B.; Charbonnier, F.; Di Fiore, F.; Tuech, J.J.; Le Pessot, F.; Michot, F.; Michel, P.; Frebourg, T. Prognostic Value of Circulating Mutant DNA in Unresectable Metastatic Colorectal Cancer. Ann. Surg. 2010, 251, 275–280. [Google Scholar] [CrossRef]
- El Messaoudi, S.; Mouliere, F.; Du Manoir, S.; Bascoul-Mollevi, C.; Gillet, B.; Nouaille, M.; Fiess, C.; Crapez, E.; Bibeau, F.; Theillet, C.G.; et al. Circulating DNA as a Strong Multimarker Prognostic Tool for Metastatic Colorectal Cancer Patient Management Care. Clin. Cancer Res. 2016, 22, 3067–3077. [Google Scholar] [CrossRef]
- Diaz, L.A., Jr.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nat. Cell Biol. 2012, 486, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nat. Cell Biol. 2012, 486, 532–536. [Google Scholar] [CrossRef]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET Receptor Drives Resistance to Anti-EGFR Therapies in Colorectal Cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef]
- Liebs, S.; Keilholz, U.; Kehler, I.; Schweiger, C.; Haybäck, J.; Nonnenmacher, A. Detection of mutations in circulating cell-free DNA in relation to disease stage in colorectal cancer. Cancer Med. 2019, 8, 3761–3769. [Google Scholar] [CrossRef] [PubMed]
- Boysen, A.; Lefevre, A.; Abrantes, R.; Johansen, J.; Jensen, B.; Schou, J.; Larsen, F.; Nielsen, D.; Taflin, H.; Gustavson, B.; et al. Methodological development and biological observations of cell free DNA with a simple direct fluorescent assay in colorectal cancer. Clin. Chim. Acta 2018, 487, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, A.; Vlajnic, M.; Ristanović, M.; Petrovic, J.; Dimitrijevic, I.; Krivokapic, Z.; Radojkovic, A. Cell-free DNA as biomarker and source for mutation detection in primary colorectal cancer. Off. J. Balk. Union Oncol. 2017, 22, 178–183. [Google Scholar]
- Santini, D.; Loupakis, F.; Vincenzi, B.; Floriani, I.; Stasi, I.; Canestrari, E.; Rulli, E.; Maltese, P.E.; Andreoni, F.; Masi, G.; et al. High Concordance ofKRASStatus Between Primary Colorectal Tumors and Related Metastatic Sites: Implications for Clinical Practice. Oncologist 2008, 13, 1270–1275. [Google Scholar] [CrossRef]
- Licar, A.; Cerkovnik, P.; Novaković, S. Distribution of some activating KRAS and BRAF mutations in Slovene patients with colorectal cancer. Med. Oncol. 2010, 28, 1048–1053. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Wickham, H. Ggplot2 Use R! Springer International Publishing: Cham, Switzerland, 2016; ISBN 978-3-319-24275-0. [Google Scholar]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2018, 47, D941–D947. [Google Scholar] [CrossRef]
- Vymetalkova, V.; Cervena, K.; Bartu, L.; Vodicka, P. Circulating Cell-Free DNA and Colorectal Cancer: A Systematic Review. Int. J. Mol. Sci. 2018, 19, 3356. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol. Detect. Quantif. 2019, 17, 100087. [Google Scholar] [CrossRef]
- Li, H.; Jing, C.; Wu, J.; Ni, J.; Sha, H.; Xu, X.; Du, Y.; Lou, R.; Dong, S.; Feng, J. Circulating tumor DNA detection: A potential tool for colorectal cancer management (Review). Oncol. Lett. 2018, 17, 1409–1416. [Google Scholar] [CrossRef] [PubMed]
- Thierry, A.R.; El Messaoudi, S.; Gahan, P.B.; Anker, P.; Stroun, M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016, 35, 347–376. [Google Scholar] [CrossRef]
- Butt, A.N.; Swaminathan, R. Overview of Circulating Nucleic Acids in Plasma/Serum. Ann. N. Y. Acad. Sci. 2008, 1137, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Zwirner, K.; Hilke, F.J.; Demidov, G.; Ossowski, S.; Gani, C.; Rieß, O.; Zips, D.; Welz, S.; Schroeder, C. Circulating cell-free DNA: A potential biomarker to differentiate inflammation and infection during radiochemotherapy. Radiother. Oncol. 2018, 129, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Peng, B.; Dong, C.; Liu, L.; Mao, J.; Wei, S.; Wang, X.; Xu, H.; Shen, J.; Mao, H.-Q.; et al. Cationic nanoparticle as an inhibitor of cell-free DNA-induced inflammation. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Breitbach, S.; Tug, S.; Simon, P. Circulating Cell-Free DNA. Sports Med. 2012, 42, 565–586. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, S.; Li, C.; Xu, C.; Zhao, J.; Miao, L. Comprehensive Evaluation of the Factors Affecting Plasma Circulating Cell-Free DNA Levels and Their Application in Diagnosing Nonsmall Cell Lung Cancer. Genet. Test. Mol. Biomarkers 2019, 23, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Lapin, M.; Oltedal, S.; Tjensvoll, K.; Buhl, T.; Smaaland, R.; Garresori, H.; Javle, M.; Glenjen, N.I.; Abelseth, B.K.; Gilje, B.; et al. Fragment size and level of cell-free DNA provide prognostic information in patients with advanced pancreatic cancer. J. Transl. Med. 2018, 16, 1–10. [Google Scholar] [CrossRef]
- Jakovljevic, K.; Malisic, E.; Cavic, M.; Krivokuca, A.; Dobricic, J.; Jankovic, R. KRAS and BRAF mutations in Serbian patients with colorectal cancer. J. B.U.ON. Off. J. Balk. Union Oncol. 2012, 17, 575–580. [Google Scholar]
- Sclafani, F.; Chau, I.; Cunningham, D.; Hahne, J.C.; Vlachogiannis, G.; Eltahir, Z.; Lampis, A.; Braconi, C.; Kalaitzaki, E.; De Castro, D.G.; et al. KRAS and BRAF mutations in circulating tumour DNA from locally advanced rectal cancer. Sci. Rep. 2018, 8, 1445. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Santini, D.; Vincenzi, B.; Addeo, R.; Garufi, C.; Masi, G.; Scartozzi, M.; Mancuso, A.; Frezza, A.M.; Venditti, O.; Imperatori, M.; et al. Cetuximab rechallenge in metastatic colorectal cancer patients: How to come away from acquired resistance? Ann. Oncol. 2012, 23, 2313–2318. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Arena, S.; Lamba, S.; Siravegna, G.; Lallo, A.; Hobor, S.; Russo, M.; Buscarino, M.; Lazzari, L.; Sartore-Bianchi, A.; et al. Blockade of EGFR and MEK Intercepts Heterogeneous Mechanisms of Acquired Resistance to Anti-EGFR Therapies in Colorectal Cancer. Sci. Transl. Med. 2014, 6, 224ra26. [Google Scholar] [CrossRef]
- Finzel, A.; Sadik, H.; Ghitti, G.; Laes, J.-F. The combined analysis of solid and liquid biopsies provides additional clinical information to improve patient care. J. Cancer Metastasis Treat. 2018, 4, 21. [Google Scholar] [CrossRef]
- Swift, S.L.; Duffy, S.; Lang, S.H. Impact of tumor heterogeneity and tissue sampling for genetic mutation testing: A systematic review and post hoc analysis. J. Clin. Epidemiol. 2020, 126, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.K.; Saller, J.; Wang, E.; Boyle, T.; Gray, J.E. Cell-free circulating tumor DNA supplementing tissue biopsies for identification of targetable mutations: Implications for precision medicine and considerations for reconciling results. Lung Cancer 2017, 111, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Tam, I.Y.S.; Chung, L.P.; Suen, W.S.; Wang, E.; Wong, M.C.; Ho, K.K.; Lam, W.K.; Chiu, S.W.; Girard, L.; Minna, J.D.; et al. Distinct Epidermal Growth Factor Receptor and KRAS Mutation Patterns in Non–Small Cell Lung Cancer Patients with Different Tobacco Exposure and Clinicopathologic Features. Clin. Cancer Res. 2006, 12, 1647–1653. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, G.; Beaty, R.; Hruban, R.H.; Maitra, A. Molecular genetics of pancreatic intraepithelial neoplasia. J. Hepato-Biliary-Pancreatic Surg. 2007, 14, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Burmer, G.C.; Loeb, L.A. Mutations in the KRAS2 oncogene during progressive stages of human colon carcinoma. Proc. Natl. Acad. Sci. USA 1989, 86, 2403–2407. [Google Scholar] [CrossRef] [PubMed]
- Bidshahri, R.; Attali, D.; Fakhfakh, K.; McNeil, K.; Karsan, A.; Won, J.R.; Wolber, R.; Bryan, J.; Hughesman, C.; Haynes, C. Quantitative Detection and Resolution of BRAF V600 Status in Colorectal Cancer Using Droplet Digital PCR and a Novel Wild-Type Negative Assay. J. Mol. Diagn. 2016, 18, 190–204. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient No. | Dukes Classification | Tumor Mutations | Serum Mutations |
|---|---|---|---|
| 1 | B | WT | WT |
| 2 | C | WT | * V600E (0.13%) |
| 3 | ND | G12D (21.10%) | G12D (0.017%) |
| 4 | B | G12D (5.50%) | G12D (0.019%) |
| 5 | C | G12V (14.66%), G12D (0.23%) | G12D (0.18%), G12V (0.036%) |
| 6 | C | G12V (5.99%) | G13D (1.95%), G12D (0.08%), G12V (0.54%), * V600E (0.09%) |
| 7 | C | G12A (19.90%), G12D (0.26%) | WT |
| 8 | C | G12V (0.018%), G13D (0.014%) | G12V (0.07%), G12D (0.046%) |
| 9 | C | G12D (0.029%) | WT |
| 10 | B | G12D (19.30%) | G12D (0.25%) |
| 11 | C | WT | WT |
| 12 | B | G12D (15.50%), *V600E (0.031%) | WT |
| 13 | B | G13D (11.75%) | G12D (0.033%) |
| 14 | C | G12V (11.28%) | WT |
| 15 | B | G12D (27.20%), G13D (0.047%) | WT |
| 16 | A | G12D (0.027%) | WT |
| 17 | B | G12V (19.90%) | G13D (7.90%), G12V (0.43%), * V600E (1.00%) |
| 18 | ND | WT | G13D (0.11%) |
| 19 | C | WT | WT |
| 20 | B | G12D (28.10%) | G12D (0.31%) |
| 21 | B | WT | G13D (0.070%) |
| 22 | ND | G12D (0.009%) | WT |
| 23 | C | G12D (0.041%) | * V600E (0.60%) |
| 24 | C | G13D (0.017%) | WT |
| 25 | B | G12D (38.90%) | WT |
| 26 | C | G12D (0.065%) | WT |
| 27 | B | WT | WT |
| 28 | C | WT | WT |
| 29 | C | WT | WT |
| 30 | ND | G12V (10.54%) | WT |
| 31 | ND | WT | WT |
| 32 | C | G13D (1.90%) | WT |
| 33 | ND | G13D (18.30%) | WT |
| 34 | A | G12V (4.84%) | WT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zmrzljak, U.P.; Košir, R.; Krivokapić, Z.; Radojković, D.; Nikolić, A. Detection of Somatic Mutations with ddPCR from Liquid Biopsy of Colorectal Cancer Patients. Genes 2021, 12, 289. https://doi.org/10.3390/genes12020289
Zmrzljak UP, Košir R, Krivokapić Z, Radojković D, Nikolić A. Detection of Somatic Mutations with ddPCR from Liquid Biopsy of Colorectal Cancer Patients. Genes. 2021; 12(2):289. https://doi.org/10.3390/genes12020289
Chicago/Turabian StyleZmrzljak, Uršula Prosenc, Rok Košir, Zoran Krivokapić, Dragica Radojković, and Aleksandra Nikolić. 2021. "Detection of Somatic Mutations with ddPCR from Liquid Biopsy of Colorectal Cancer Patients" Genes 12, no. 2: 289. https://doi.org/10.3390/genes12020289
APA StyleZmrzljak, U. P., Košir, R., Krivokapić, Z., Radojković, D., & Nikolić, A. (2021). Detection of Somatic Mutations with ddPCR from Liquid Biopsy of Colorectal Cancer Patients. Genes, 12(2), 289. https://doi.org/10.3390/genes12020289