
Current Status of Genetic Diagnosis Laboratories and Frequency of Genetic Variants Associated with Cystic Fibrosis through a Newborn-Screening Program in Turkey

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Sampling
2.2. Next-Generation Sequencing (NGS)
2.3. Bioinformatics Analyses
2.4. Multiplex Ligation-Dependent Probe Amplification (MLPA) Analyses
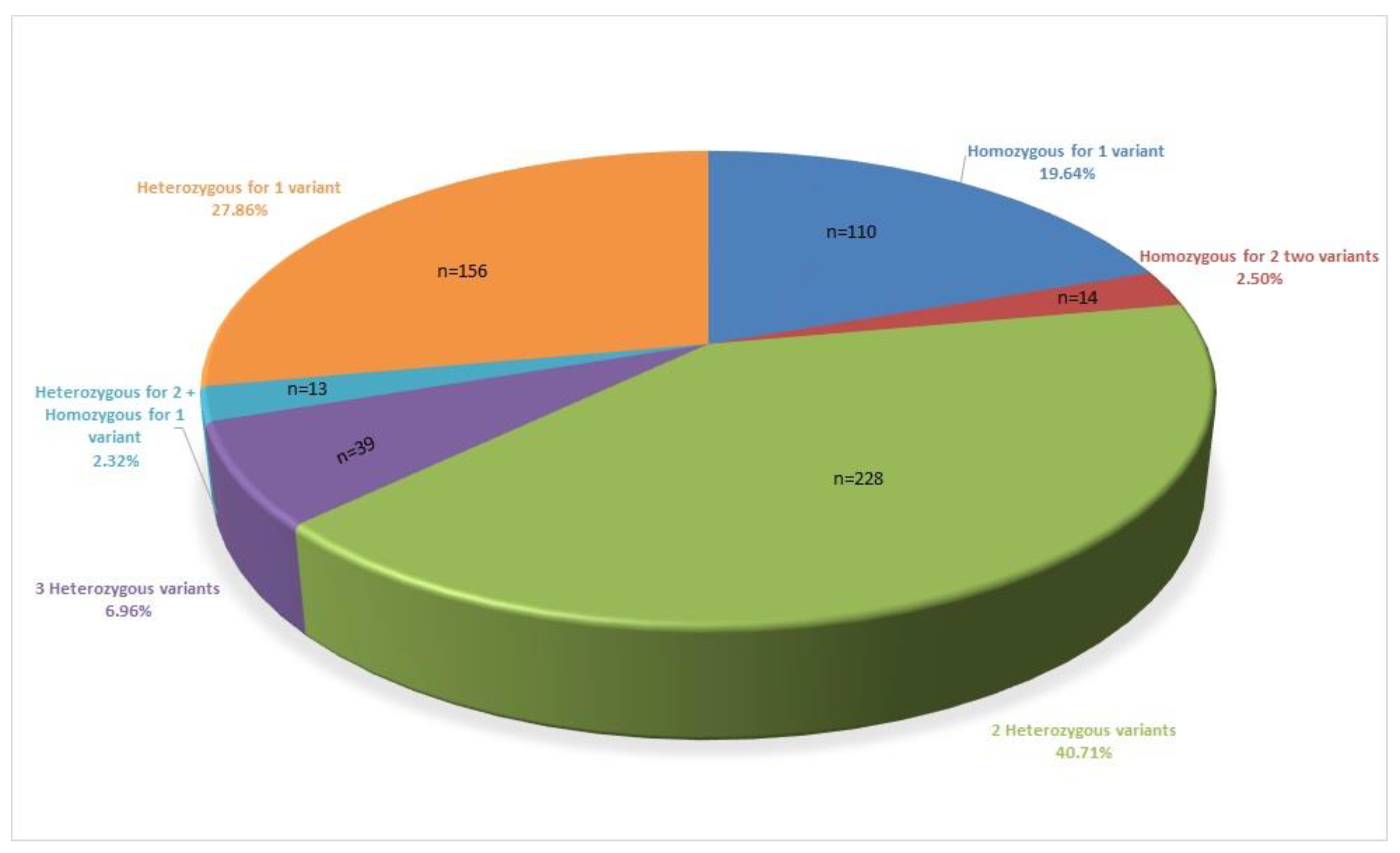
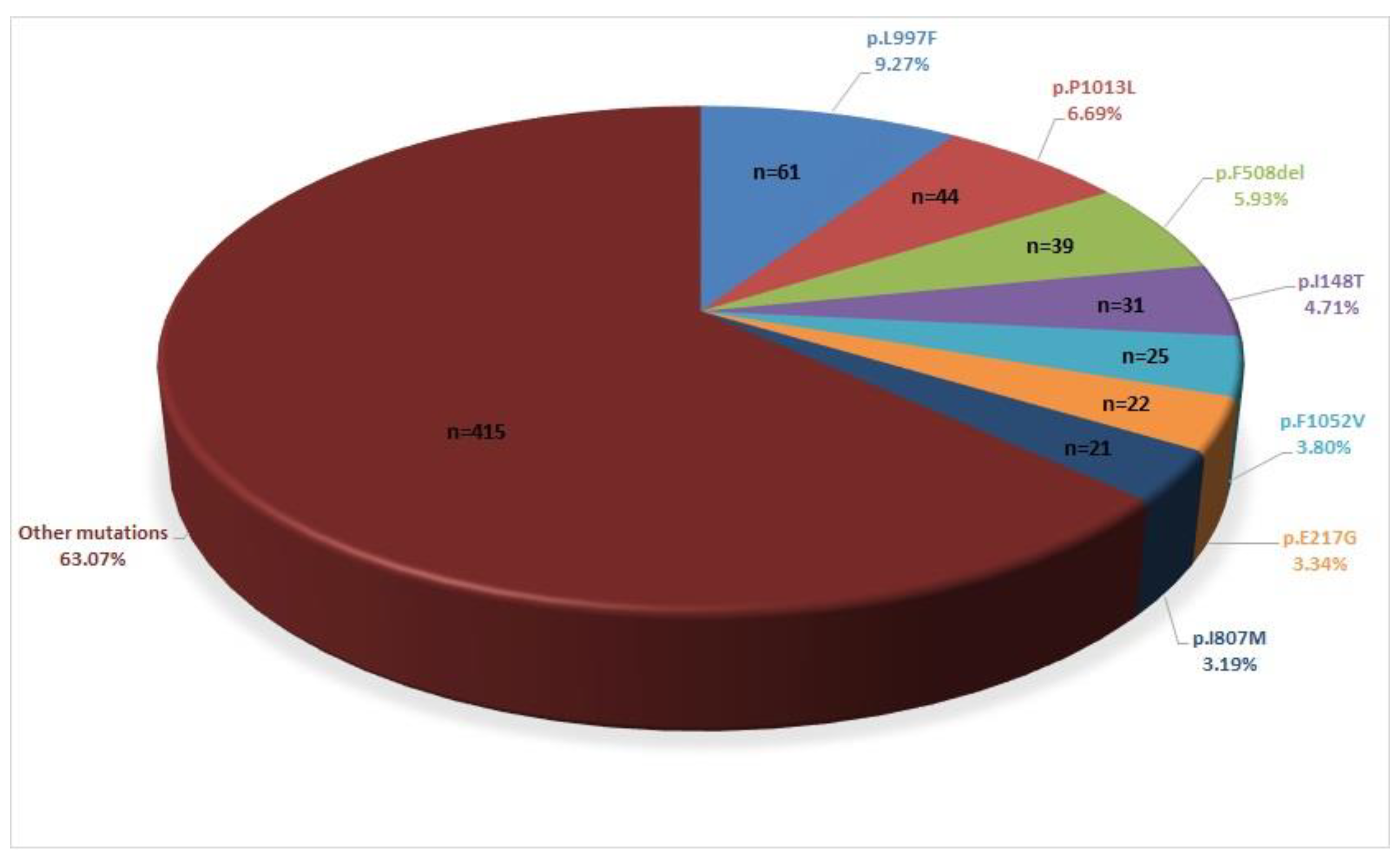
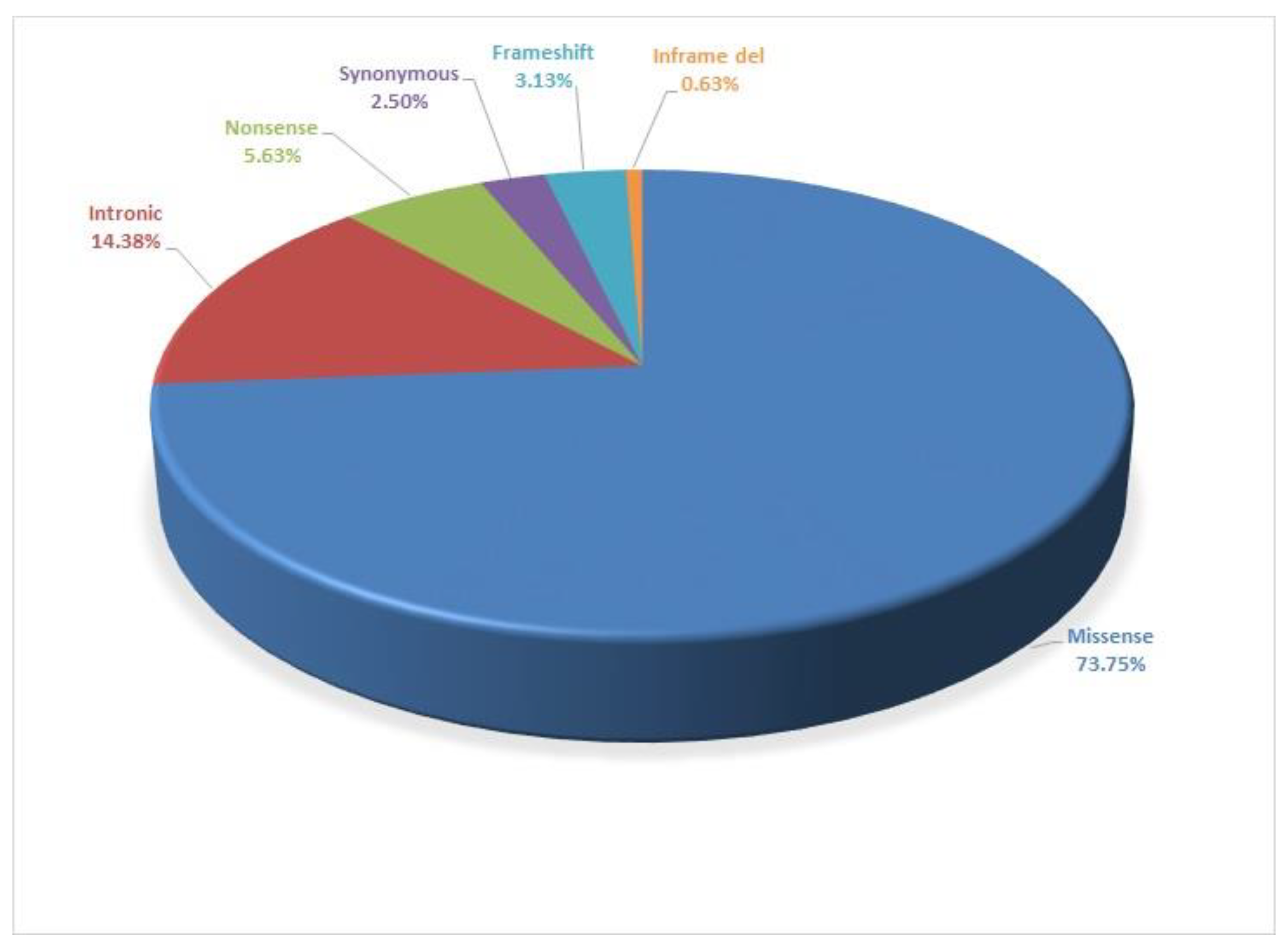
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schrijver, I. Mutation Distribution in Expanded Screening for Cystic Fibrosis: Making Up the Balance in a Context of Ethnic Diversity. Clin. Chem. 2011, 57, 799–801. [Google Scholar] [CrossRef] [PubMed][Green Version]
- World Health Organization. The Molecular Genetic Epidemiology of Cystic Fibrosis. Available online: https://www.who.int/genomics/publications/en/HGN_WB_04.02_report.pdf?ua=1 (accessed on 13 December 2020).
- Lago, J.E.F.; Cayarga, A.A.; González, Y.J.G.; Mesa, T.C. A simple, fast and inexpensive method for mutation scanning of CFTR gene. BMC Med. Genet. 2017, 18, 58. [Google Scholar] [CrossRef] [PubMed]
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N.; et al. Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science 1989, 245, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wrennall, J.A.; Cai, Z.; Li, H.; Sheppard, D.N. Understanding how cystic fibrosis mutations disrupt CFTR function: From single molecules to animal models. Int. J. Biochem. Cell Biol. 2014, 52, 47–57. [Google Scholar] [CrossRef]
- Human Gene Mutation Database. Available online: http://www.hgmd.cf.ac.uk/ac/gene.php?gene=CFTR (accessed on 13 December 2020).
- Svensson, A.M.; Chou, L.S.; Miller, C.E.; Robles, J.A.; Swensen, J.J.; Voelkerding, K.V.; Mao, R.; Lyon, E. Detection of large rearrangements in the cystic fibrosis transmembrane conductance regulator gene by multiplex ligation-dependent probe amplification assay when sequencing fails to detect two disease-causing mutations. Genet. Test. Mol. Biomark. 2010, 14, 171–174. [Google Scholar] [CrossRef]
- Mall, M.A.; Hartl, D. CFTR: Cystic fibrosis and beyond. Eur. Respir. J. 2014, 44, 1042. [Google Scholar] [CrossRef]
- Cystic Fibrosis Foundation. Cystic Fibrosis Foundation Patient Registry 2018 Annual Data Report. ©2020; Cystic Fibrosis Foundation: Bethesda, MD, USA, 2019. [Google Scholar]
- De Boeck, K.; Zolin, A.; Cuppens, H.; Olesen, H.V.; Viviani, L. The relative frequency of CFTR mutation classes in European patients with cystic fibrosis. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2014, 13, 403–409. [Google Scholar] [CrossRef]
- TÜİK. Available online: https://data.tuik.gov.tr/Bulten/Index?p=Dogum-Istatistikleri-2019-33706 (accessed on 14 December 2020).
- Şaşihüseyinoğlu, A.; Altıntaş, D.U.; Bişgin, A.; Doğruel, D.; Yılmaz, M.; Serbes, M. Two years of newborn screening for cystic fibrosis in Turkey: Çukurova experience. Turk. J. Pediatrics 2019, 61, 505–512. [Google Scholar] [CrossRef]
- Nefzi, M.; Hadj Fredj, S.; Tebib, N.; Barsaoui, S.; Boussetta, K.; Siala, H.; Messaoud, T. Contribution of M470V variant to cystic fibrosis: First study in CF and normal Tunisian population. Pathol. Biol. 2015, 63, 169–174. [Google Scholar] [CrossRef]
- Cutting, G.R. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat. Rev. Genet. 2015, 16, 45–56. [Google Scholar] [CrossRef]
- Dequeker, E.; Stuhrmann, M.; Morris, M.A.; Casals, T.; Castellani, C.; Claustres, M.; Cuppens, H.; des Georges, M.; Ferec, C.; Macek, M.; et al. Best practice guidelines for molecular genetic diagnosis of cystic fibrosis and CFTR-related disorders—Updated European recommendations. Eur. J. Hum. Genet. 2009, 17, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Bobadilla, J.L.; Macek, M., Jr.; Fine, J.P.; Farrell, P.M. Cystic fibrosis: A worldwide analysis of CFTR mutations—Correlation with incidence data and application to screening. Hum. Mutat. 2002, 19, 575–606. [Google Scholar] [CrossRef] [PubMed]
- Petrova, G.; Yaneva, N.; Hrbková, J.; Libik, M.; Savov, A.; Macek, M., Jr. Identification of 99% of CFTR gene mutations in Bulgarian-, Bulgarian Turk-, and Roma cystic fibrosis patients. Mol. Genet. Genom. Med. 2019, 7, e696. [Google Scholar] [CrossRef]
- American College of Obstetricians and Gynecologists; American College of Medical Genetics. Preconception and Prenatal Carrier Screening for Cystic Fibrosis: Clinical and Laboratory Guidelines; American College of Obstetricians and Gynecologists: Washington, DC, USA; American College of Medical Genetics: Bethesda, MD, USA, 2001. [Google Scholar]
- Petrova, N.V.; Kashirskaya, N.Y.; Vasilyeva, T.A.; Kondratyeva, E.I.; Zhekaite, E.K.; Voronkova, A.Y.; Sherman, V.D.; Galkina, V.A.; Ginter, E.K.; Kutsev, S.I.; et al. Analysis of CFTR Mutation Spectrum in Ethnic Russian Cystic Fibrosis Patients. Genes 2020, 11, 554. [Google Scholar] [CrossRef] [PubMed]
- Strom, C.M.; Redman, J.B.; Peng, M. The dangers of including nonclassical cystic fibrosis variants in population-based screening panels: P.L997F, further genotype/phenotype correlation data. Genet. Med. Off. J. Am. Coll. Med. Genet. 2011, 13, 1042–1044. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, M.; Narzi, L.; Pierandrei, S.; Bruno, S.M.; Stamato, A.; d’Avanzo, M.; Strom, R.; Quattrucci, S. A new complex allele of the CFTR gene partially explains the variable phenotype of the L997F mutation. Genet. Med. Off. J. Am. Coll. Med. Genet. 2010, 12, 548–555. [Google Scholar] [CrossRef] [PubMed]
- LaRusch, J.; Jung, J.; General, I.J.; Lewis, M.D.; Park, H.W.; Brand, R.E.; Gelrud, A.; Anderson, M.A.; Banks, P.A.; Conwell, D.; et al. Mechanisms of CFTR functional variants that impair regulated bicarbonate permeation and increase risk for pancreatitis but not for cystic fibrosis. PLoS Genet 2014, 10, e1004376. [Google Scholar] [CrossRef] [PubMed]
- Sofia, V.M.; Surace, C.; Terlizzi, V.; Da Sacco, L.; Alghisi, F.; Angiolillo, A.; Braggion, C.; Cirilli, N.; Colombo, C.; Di Lullo, A.; et al. Trans-heterozygosity for mutations enhances the risk of recurrent/chronic pancreatitis in patients with Cystic Fibrosis. Mol. Med. 2018, 24, 38. [Google Scholar] [CrossRef] [PubMed]
- Terlizzi, V.; Castaldo, G.; Salvatore, D.; Lucarelli, M.; Raia, V.; Angioni, A.; Carnovale, V.; Cirilli, N.; Casciaro, R.; Colombo, C.; et al. Genotype-phenotype correlation and functional studies in patients with cystic fibrosis bearing CFTR complex alleles. J. Med. Genet. 2017, 54, 224–235. [Google Scholar] [CrossRef]
- Derichs, N.; Schuster, A.; Grund, I.; Ernsting, A.; Stolpe, C.; Körtge-Jung, S.; Gallati, S.; Stuhrmann, M.; Kozlowski, P.; Ballmann, M. Homozygosity for L997F in a child with normal clinical and chloride secretory phenotype provides evidence that this cystic fibrosis transmembrane conductance regulator mutation does not cause cystic fibrosis. Clin. Genet. 2005, 67, 529–531. [Google Scholar] [CrossRef]
- Taccetti, G.; Botti, M.; Terlizzi, V.; Cavicchi, M.C.; Neri, A.S.; Galici, V.; Mergni, G.; Centrone, C.; Peroni, D.G.; Festini, F. Clinical and Genotypical Features of False-Negative Patients in 26 Years of Cystic Fibrosis Neonatal Screening in Tuscany, Italy. Diagnostics 2020, 10, 446. [Google Scholar] [CrossRef] [PubMed]
- Suaud, L.; Yan, W.; Rubenstein, R.C. Abnormal regulatory interactions of I148T-CFTR and the epithelial Na+ channel in Xenopus oocytes. Am. J. Physiol. Cell Physiol. 2007, 292, C603–C611. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, K.G.; Highsmith, W.E.; Amos, J.; Pratt, V.M.; Roa, B.; Friez, M.; Pike-Buchanan, L.L.; Buyse, I.M.; Redman, J.B.; Strom, C.M.; et al. Genotype-phenotype correlation and frequency of the 3199del6 cystic fibrosis mutation among I148T carriers: Results from a collaborative study. Genet. Med. Off. J. Am. Coll. Med. Genet. 2004, 6, 421–425. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Loumi, O.; Cuppens, H.; Bakour, R.; Benabadji, M.; Baghriche, M.; Marynen, P.; Cassiman, J.J. An Algerian child homozygous for the M470V polymorphism and for a deletion of two nucleotides in exon 10 of the CFTR gene, shows severe cystic fibrosis symptoms. Genet. Couns. 1992, 3, 205–207. [Google Scholar] [PubMed]
- Veit, G.; Da Fonte, D.F.; Avramescu, R.G.; Premchandar, A.; Bagdany, M.; Xu, H.; Bensinger, D.; Stubba, D.; Schmidt, B.; Matouk, E.; et al. Mutation-specific dual potentiators maximize rescue of CFTR gating mutants. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2020, 19, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Banjar, H.H.; Tuleimat, L.; El Seoudi, A.A.A.; Mogarri, I.; Alhaider, S.; Nizami, I.Y.; AlMaghamsi, T.; Alkaf, S.A.; Moghrabi, N. Genotype patterns for mutations of the cystic fibrosis transmembrane conductance regulator gene: A retrospective descriptive study from Saudi Arabia. Ann. Saudi Med. 2020, 40, 15–24. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Variants | Mutation Type | ACMG Classification | Patient Numbers |
|---|---|---|---|
| c.2978A > C p.D993A | Missense | LP | 7 |
| c.1729T > C p.Y577H | Missense | VUS | 2 |
| c.3468 + 52A > C | Intronic | VUS | 2 |
| c.3170C > G p.T1057R | Missense | LP | 1 |
| c.2982C > G p.F994L | Missense | VUS | 1 |
| c.1680 − 756C > T | Intronic | VUS | 1 |
| c.2054A > G p.Q685R | Missense | LP | 1 |
| c.698T > C p.L233P | Missense | LP | 1 |
| c.4321delC p.R1438fs*10 | Frameshift | LP | 1 |
| c.3468 + 137T > C | Intronic | VUS | 1 |
| c.7A > T p.R3W | Missense | VUS | 1 |
| c.1836A > T p.K612N | Missense | LP | 1 |
| c.1217G > C p.G406A | Missense | VUS | 1 |
| c.53 + 28A > G | Intronic | VUS | 1 |
| c.3311A > G p.E1104G | Missense | LP | 1 |
| Variants | Mutation Type | Number of Alleles | ACMG Classification | CFRT2 Database |
|---|---|---|---|---|
| p.V470M | Missense | 399 | Benign * | N/A |
| p.L997F | Missense | 66 | P | Non CF-causing |
| p.P1013L | Missense | 47 | P | N/A |
| p.F508del | Inframe del | 42 | P | CF-causing |
| p.I148T | Missense | 31 | VUS | Non CF-causing |
| p.F1052V | Missense | 25 | P | Varying clinical consequence |
| p.E217G | Missense | 22 | P | N/A |
| p.I807M | Missense | 22 | P | Non CF-causing |
| c.2657 + 5G > A | Intronic | 19 | P | CF-causing |
| c.2620 − 15C > G | Intronic | 16 | P | N/A |
| c.3964 − 3C > G | Intronic | 16 | P | N/A |
| p.F1052L | Missense | 13 | P | N/A |
| p.K68E | Missense | 13 | P | N/A |
| p.D1312G | Missense | 11 | P | N/A |
| p.D993A | Missense | 9 | LP | N/A |
| p.M952I | Missense | 9 | P | N/A |
| p.Y515 * | Nonsense | 9 | P | N/A |
| c.2909 − 71G > C | Intronic | 8 | P | N/A |
| c.3718 − 2477C > T | Intronic | 8 | P | CF-causing |
| p.D110H | Missense | 8 | P | CF-causing |
| p.D1152H | Missense | 8 | P | Varying clinical consequence |
| p.K684fs*38 | Frameshift | 8 | P | N/A |
| p.M348K | Missense | 8 | P | N/A |
| p.W1282 * | Nonsense | 8 | P | CF-causing |
| p.D806G | Missense | 7 | P | N/A |
| p.G576A | Missense | 7 | VUS | Non CF-causing |
| p.L732 * | Nonsense | 7 | P | CF-causing |
| p.N1303K | Missense | 7 | P | CF-causing |
| p.R347P | Missense | 7 | P | CF-causing |
| p.I506V | Missense | 6 | VUS | N/A |
| p.L633I | Missense | 6 | P | N/A |
| p.R117C | Missense | 6 | P | CF-causing |
| p.T388M | Missense | 6 | P | N/A |
| c.489 + 3A > G | Intronic | 5 | P | Varying clinical consequence |
| p.G542 * | Nonsense | 5 | P | Varying clinical consequence |
| p.R668C | Missense | 5 | P | Non CF-causing |
| p.R75Q | Missense | 5 | P | Non CF-causing |
| p.S1235R | Missense | 5 | P | Non CF-causing |
| p.S877A | Missense | 5 | VUS | N/A |
| p.T1220I | Missense | 5 | VUS | N/A |
| p.D513G | Missense | 4 | P | CF-causing |
| p.L1034F | Missense | 4 | P | N/A |
| p.P111L | Missense | 4 | P | N/A |
| p.Q493P | Missense | 4 | P | N/A |
| p.R117H | Missense | 4 | P | Varying clinical consequence |
| p.R297Q | Missense | 4 | P | N/A |
| p.W1098C | Missense | 4 | VUS | CF-causing |
| p.A120T | Missense | 3 | LP | Varying clinical consequence |
| p.A399V | Missense | 3 | P | N/A |
| p.D836Y | Missense | 3 | P | Non CF-causing |
| p.E1228G | Missense | 3 | LP | N/A |
| p.E528K | Missense | 3 | LP | N/A |
| p.E831 * | Nonsense | 3 | P | CF-causing |
| p.E92K | Missense | 3 | P | CF-causing |
| p.I1234V | Missense | 3 | P | CF-causing |
| p.I853I | Synonymous | 3 | VUS | N/A |
| p.L183I | Missense | 3 | P | N/A |
| p.M1101R | Missense | 3 | P | CF-causing |
| p.Q353 * | Nonsense | 3 | P | N/A |
| p.R334W | Missense | 3 | P | CF-causing |
| p.R352Q | Missense | 3 | VUS | CF-causing |
| p.S955A | Missense | 3 | VUS | N/A |
| p.V1198M | Missense | 3 | LP | N/A |
| c.1766 + 3A > G | Intronic | 2 | P | CF-causing |
| c.2491 − 51T > C | Intronic | 2 | VUS | N/A |
| c.3468 + 52A > C | Intronic | 2 | VUS | N/A |
| p.C866T | Missense | 2 | LP | N/A |
| p.E528E | Missense | 2 | P | N/A |
| p.F834L | Missense | 2 | P | N/A |
| p.G1069R | Missense | 2 | P | Varying clinical consequence |
| p.I1000fs*2 | Frameshift | 2 | P | N/A |
| p.I1295fs *33 | Frameshift | 2 | P | N/A |
| p.I752S | Missense | 2 | VUS | N/A |
| p.K64E | Missense | 2 | P | N/A |
| p.L137fs*15 | Frameshift | 2 | P | N/A |
| p.M952T | Missense | 2 | P | Unknown significance |
| p.R709 * | Nonsense | 2 | P | CF-causing |
| p.R74W | Missense | 2 | P | Varying clinical consequence |
| p.S1373I | Missense | 2 | LP | N/A |
| p.T1019A | Missense | 2 | VUS | N/A |
| p.T1057A | Missense | 2 | P | N/A |
| p.T1299T | Missense | 2 | VUS | N/A |
| p.T966M | Missense | 2 | VUS | N/A |
| p.V201M | Missense | 2 | P | Unknown significance |
| p.V754M | Missense | 2 | P | Non CF-causing |
| p.Y301C | Missense | 2 | VUS | N/A |
| p.Y577H | Missense | 2 | VUS | N/A |
| c.1116 + 57C > G | Intronic | 1 | VUS | N/A |
| c.164 + 9A > T | Intronic | 1 | VUS | N/A |
| c.1680 − 756C > T | Intronic | 1 | VUS | N/A |
| c.2490+5G > T | Intronic | 1 | VUS | N/A |
| c.2909 − 15T > G | Intronic | 1 | VUS | N/A |
| c.2989 − 3C>T | Intronic | 1 | VUS | N/A |
| c.3139 + 80delA | Intronic | 1 | VUS | N/A |
| c.3368 − 4A > G | Intronic | 1 | P | N/A |
| c.3468 + 137T > C | Intronic | 1 | VUS | N/A |
| c.3469 − 2A > G | Intronic | 1 | P | N/A |
| c.3963+15T > C | Intronic | 1 | VUS | N/A |
| c.490 − 165T > C | Intronic | 1 | VUS | N/A |
| c.53+28A > G | Intronic | 1 | VUS | N/A |
| c.870 − 1026delC | Intronic | 1 | VUS | N/A |
| p.A1009T | Missense | 1 | P | N/A |
| p.A1113V | Missense | 1 | VUS | N/A |
| p.A1364A | Synonymous | 1 | P | N/A |
| p.A455V | Missense | 1 | P | N/A |
| p.D58G | Missense | 1 | P | N/A |
| p.D891G | Missense | 1 | P | N/A |
| p.D924N | Missense | 1 | P | Unknown significance |
| p.D985E | Missense | 1 | LP | N/A |
| p.E1104G | Missense | 1 | LP | N/A |
| p.E1409K | Missense | 1 | P | N/A |
| p.E826K | Missense | 1 | P | N/A |
| p.F508C | Missense | 1 | P | Non CF-causing |
| p.F994C | Missense | 1 | P | N/A |
| p.F994L | Missense | 1 | VUS | N/A |
| p.G314A | Missense | 1 | LP | N/A |
| p.G406A | Missense | 1 | VUS | N/A |
| p.G723D | Missense | 1 | LP | N/A |
| p.I125T | Missense | 1 | P | N/A |
| p.I521F | Missense | 1 | P | N/A |
| p.K1060T | Missense | 1 | P | N/A |
| p.K536E | Missense | 1 | LP | N/A |
| p.K612N | Missense | 1 | LP | N/A |
| p.K68N | Missense | 1 | VUS | N/A |
| p.L1156F | Missense | 1 | VUS | N/A |
| p.L227R | Missense | 1 | P | CF-causing |
| p.L233P | Missense | 1 | LP | N/A |
| p.L467F | Missense | 1 | P | N/A |
| p.L568F | Missense | 1 | VUS | N/A |
| p.L610I | Missense | 1 | P | N/A |
| p.L88 * | Nonsense | 1 | P | CF-causing |
| p.M1407T | Missense | 1 | P | N/A |
| p.N1432K | Missense | 1 | P | N/A |
| p.N306S | Missense | 1 | VUS | N/A |
| p.N417K | Missense | 1 | VUS | N/A |
| p.P111P | Synonymous | 1 | VUS | N/A |
| p.P5L | Missense | 1 | P | Varying clinical consequence |
| p.Q685R | Missense | 1 | LP | N/A |
| p.R1162L | Missense | 1 | P | Non CF-causing |
| p.R1438fs*10 | Frameshift | 1 | LP | N/A |
| p.R31C | Missense | 1 | P | Non CF-causing |
| p.R334Q | Missense | 1 | P | Varying clinical consequence |
| p.R347H | Missense | 1 | P | CF-causing |
| p.R3W | Missense | 1 | VUS | N/A |
| p.R668L | Missense | 1 | P | N/A |
| p.R785 * | Nonsense | 1 | P | CF-causing |
| p.R997F | Missense | 1 | P | N/A |
| p.S1426P | Missense | 1 | P | N/A |
| p.S307N | Missense | 1 | P | N/A |
| p.S912L | Missense | 1 | P | Unknown significance |
| p.S945L | Missense | 1 | P | CF-causing |
| p.T1057R | Missense | 1 | LP | N/A |
| p.T351S | Missense | 1 | P | N/A |
| p.T465N | Missense | 1 | P | N/A |
| p.T760M | Missense | 1 | P | N/A |
| p.V855I | Missense | 1 | VUS | N/A |
| p.V920L | Missense | 1 | P | N/A |
| p.V938G | Missense | 1 | LP | N/A |
| p.W1098L | Missense | 1 | P | N/A |
| p.Y424Y | Synonymous | 1 | P | N/A |
| p.Y919C | Missense | 1 | P | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bozdogan, S.T.; Mujde, C.; Boga, I.; Sonmezler, O.; Hanta, A.; Rencuzogullari, C.; Ozcan, D.; Altintas, D.U.; Bisgin, A. Current Status of Genetic Diagnosis Laboratories and Frequency of Genetic Variants Associated with Cystic Fibrosis through a Newborn-Screening Program in Turkey. Genes 2021, 12, 206. https://doi.org/10.3390/genes12020206
Bozdogan ST, Mujde C, Boga I, Sonmezler O, Hanta A, Rencuzogullari C, Ozcan D, Altintas DU, Bisgin A. Current Status of Genetic Diagnosis Laboratories and Frequency of Genetic Variants Associated with Cystic Fibrosis through a Newborn-Screening Program in Turkey. Genes. 2021; 12(2):206. https://doi.org/10.3390/genes12020206
Chicago/Turabian StyleBozdogan, Sevcan Tug, Cem Mujde, Ibrahim Boga, Ozge Sonmezler, Abdullah Hanta, Cagla Rencuzogullari, Dilek Ozcan, Derya Ufuk Altintas, and Atil Bisgin. 2021. "Current Status of Genetic Diagnosis Laboratories and Frequency of Genetic Variants Associated with Cystic Fibrosis through a Newborn-Screening Program in Turkey" Genes 12, no. 2: 206. https://doi.org/10.3390/genes12020206
APA StyleBozdogan, S. T., Mujde, C., Boga, I., Sonmezler, O., Hanta, A., Rencuzogullari, C., Ozcan, D., Altintas, D. U., & Bisgin, A. (2021). Current Status of Genetic Diagnosis Laboratories and Frequency of Genetic Variants Associated with Cystic Fibrosis through a Newborn-Screening Program in Turkey. Genes, 12(2), 206. https://doi.org/10.3390/genes12020206