
On the Origin and Evolution of Drosophila New Genes during Spermatogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
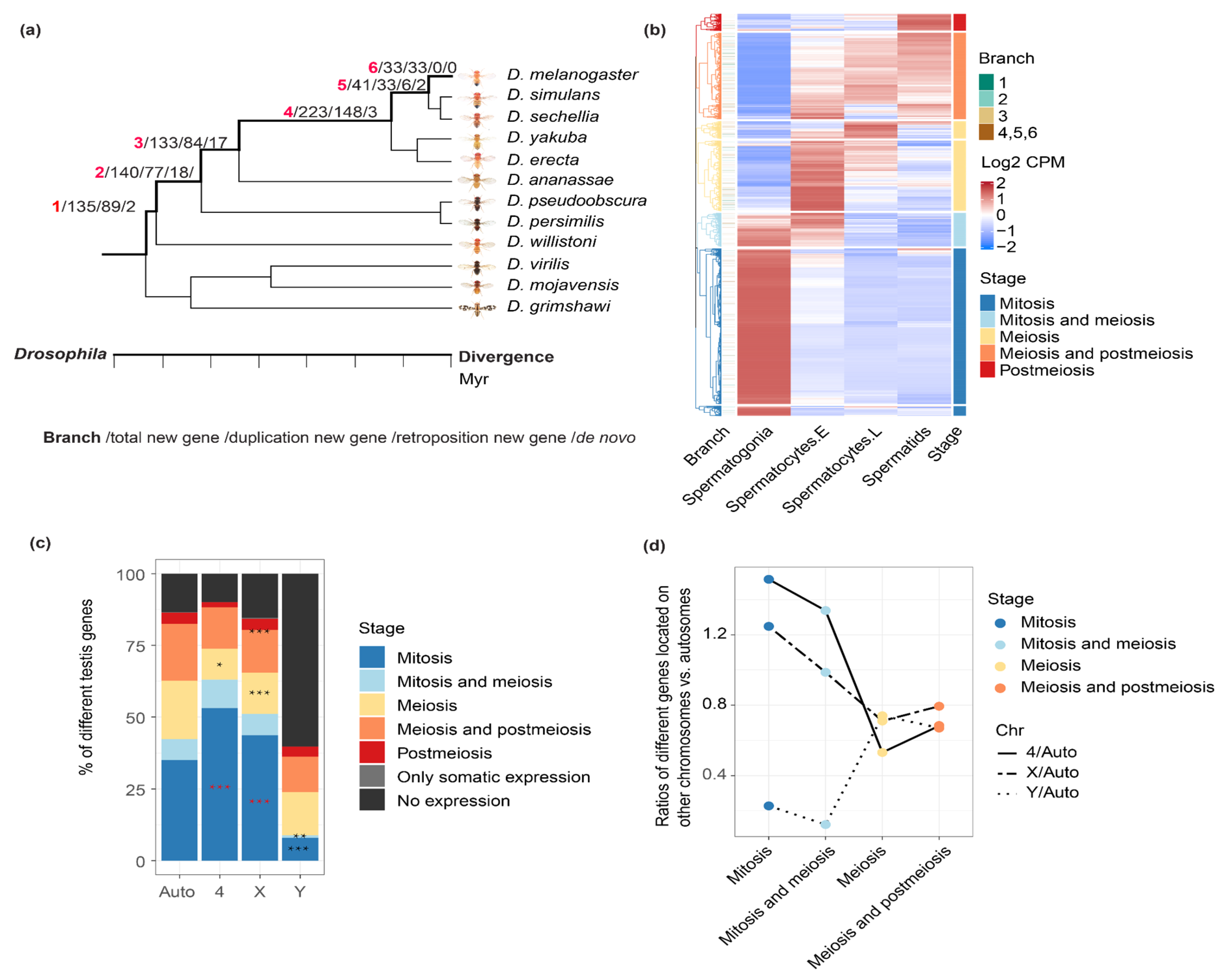
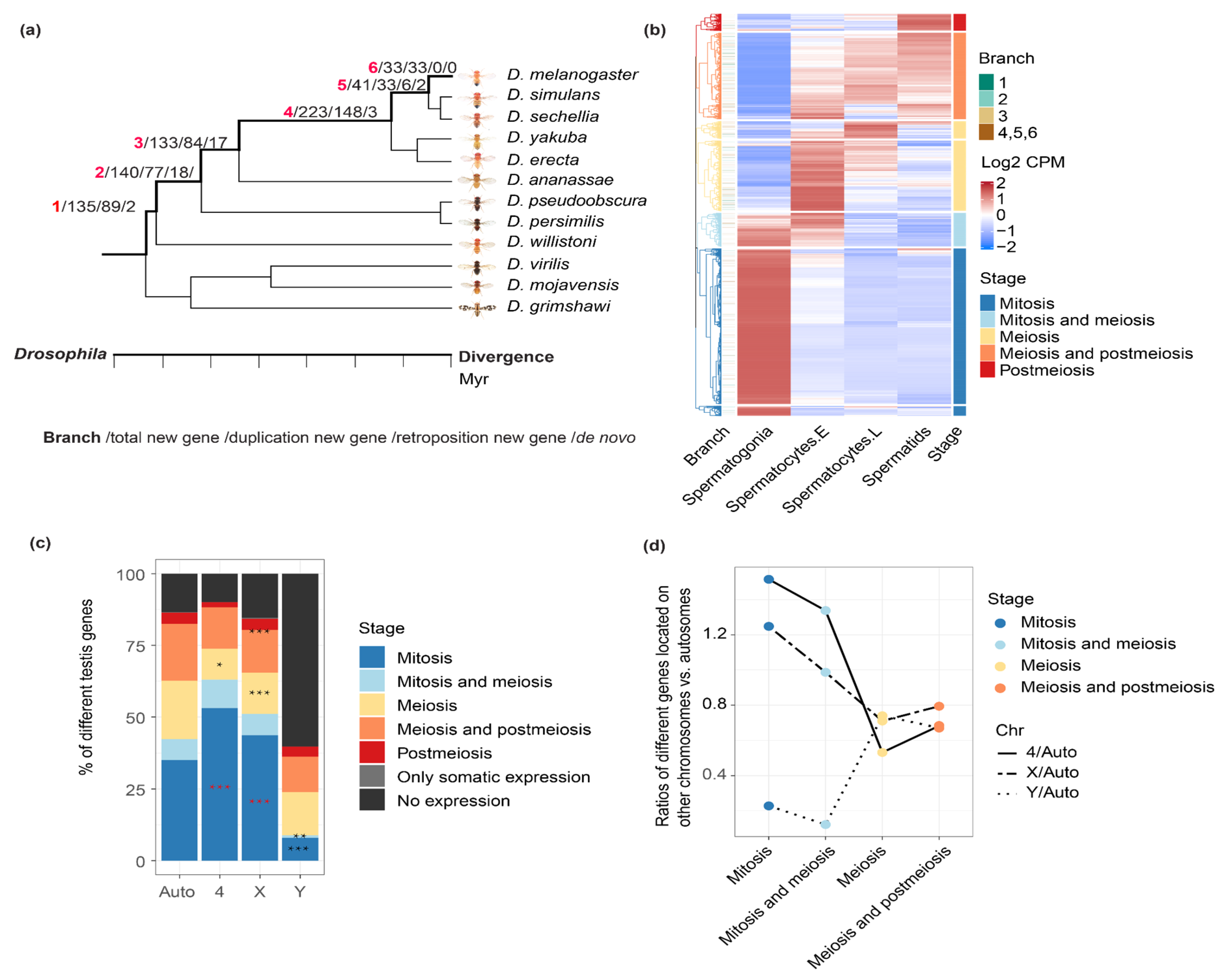
2.1. Comparing the Expression Patterns of Genes between Chromosomes during Spermatogenesis
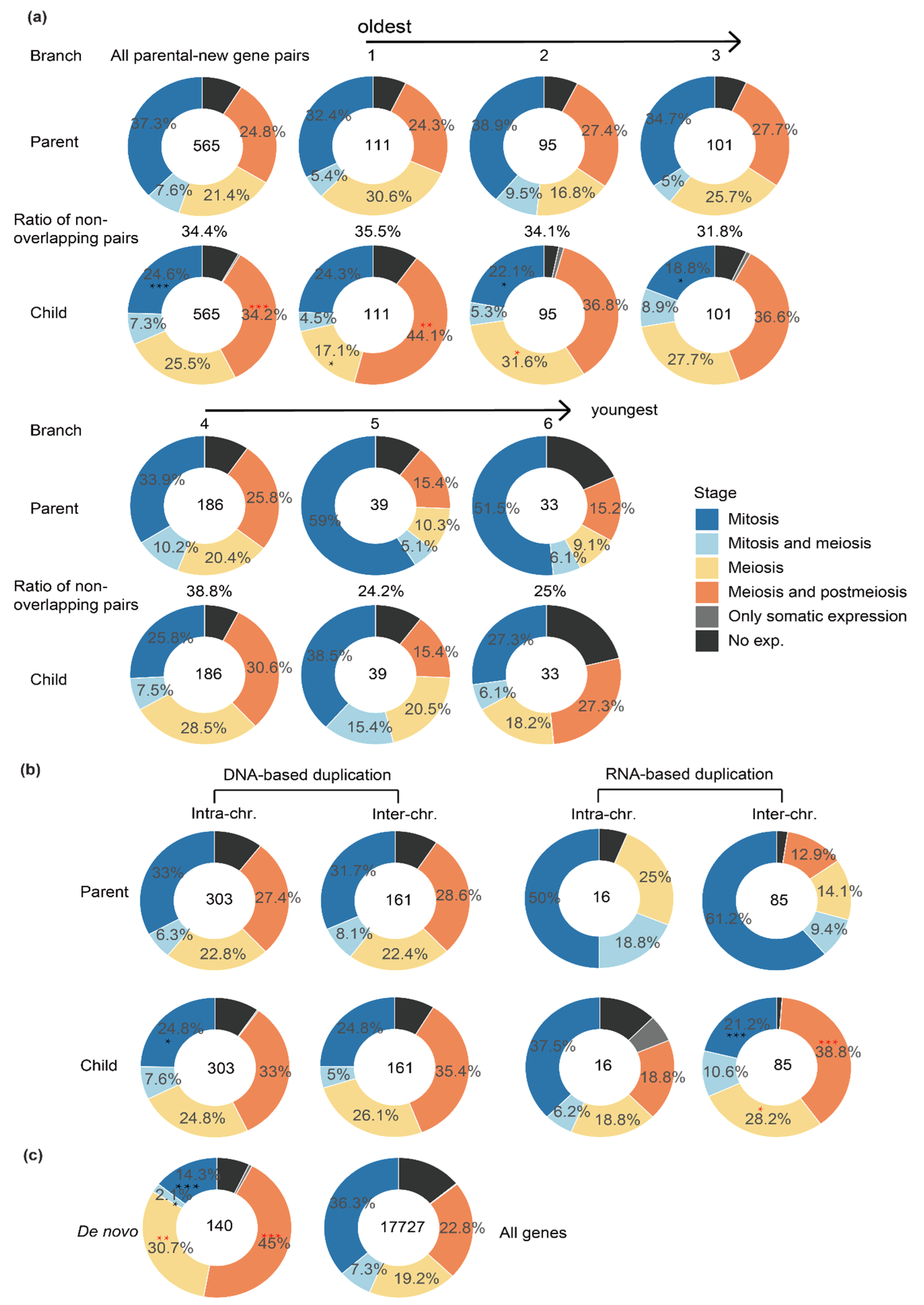
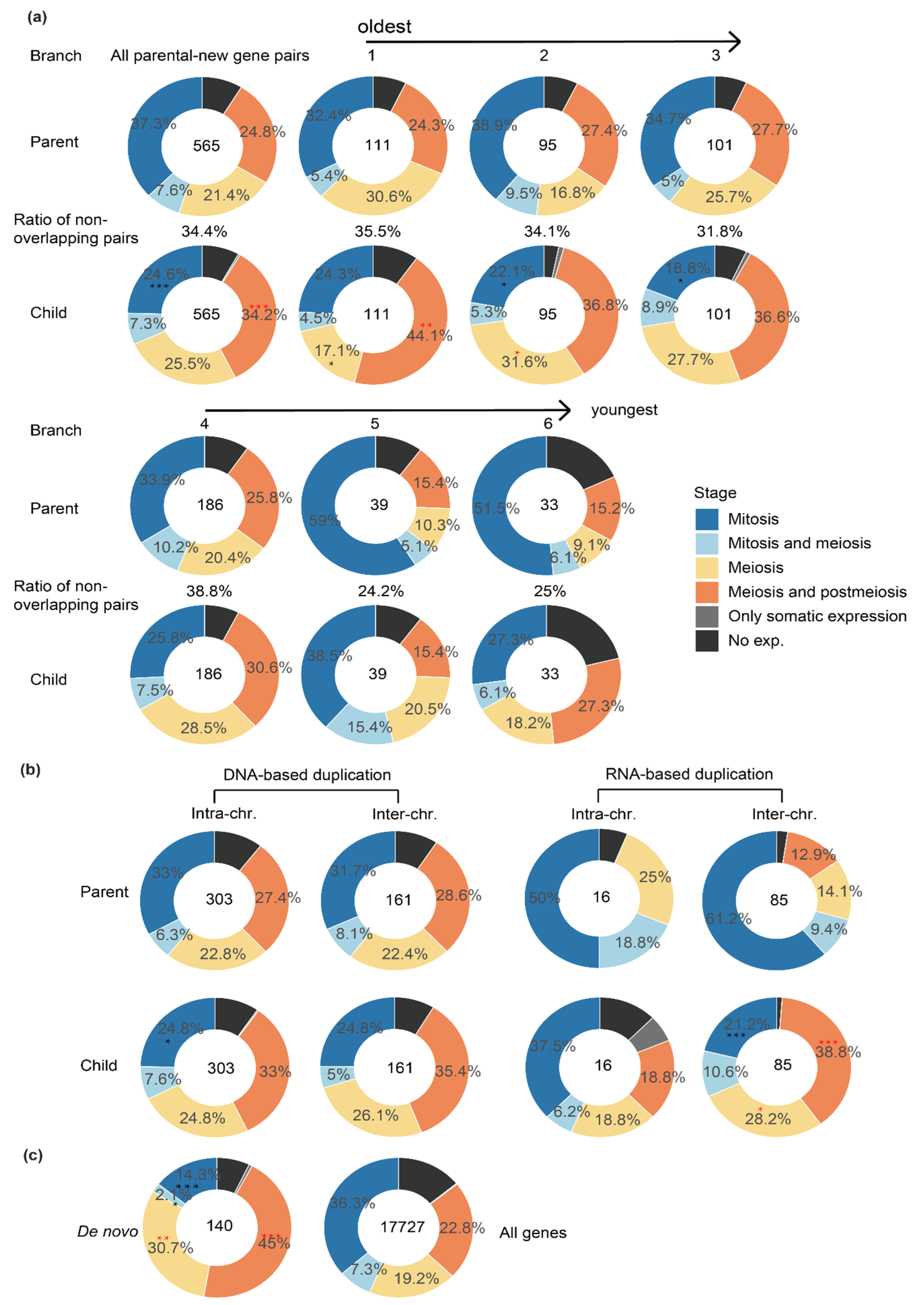
2.2. New Genes of Different Ages or Produced by Different Mechanisms Have a Different Expression Pattern across Testis Cells
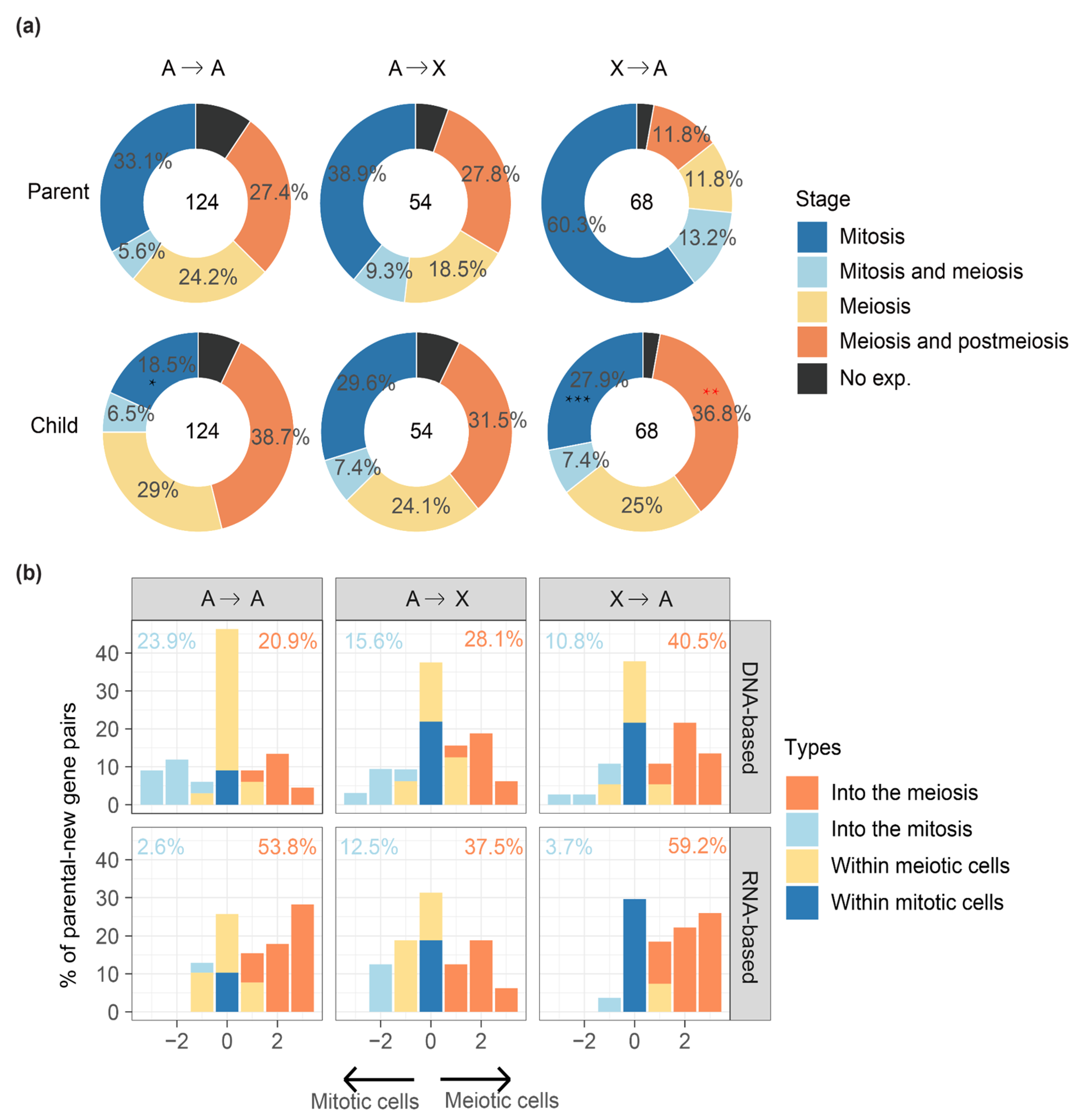
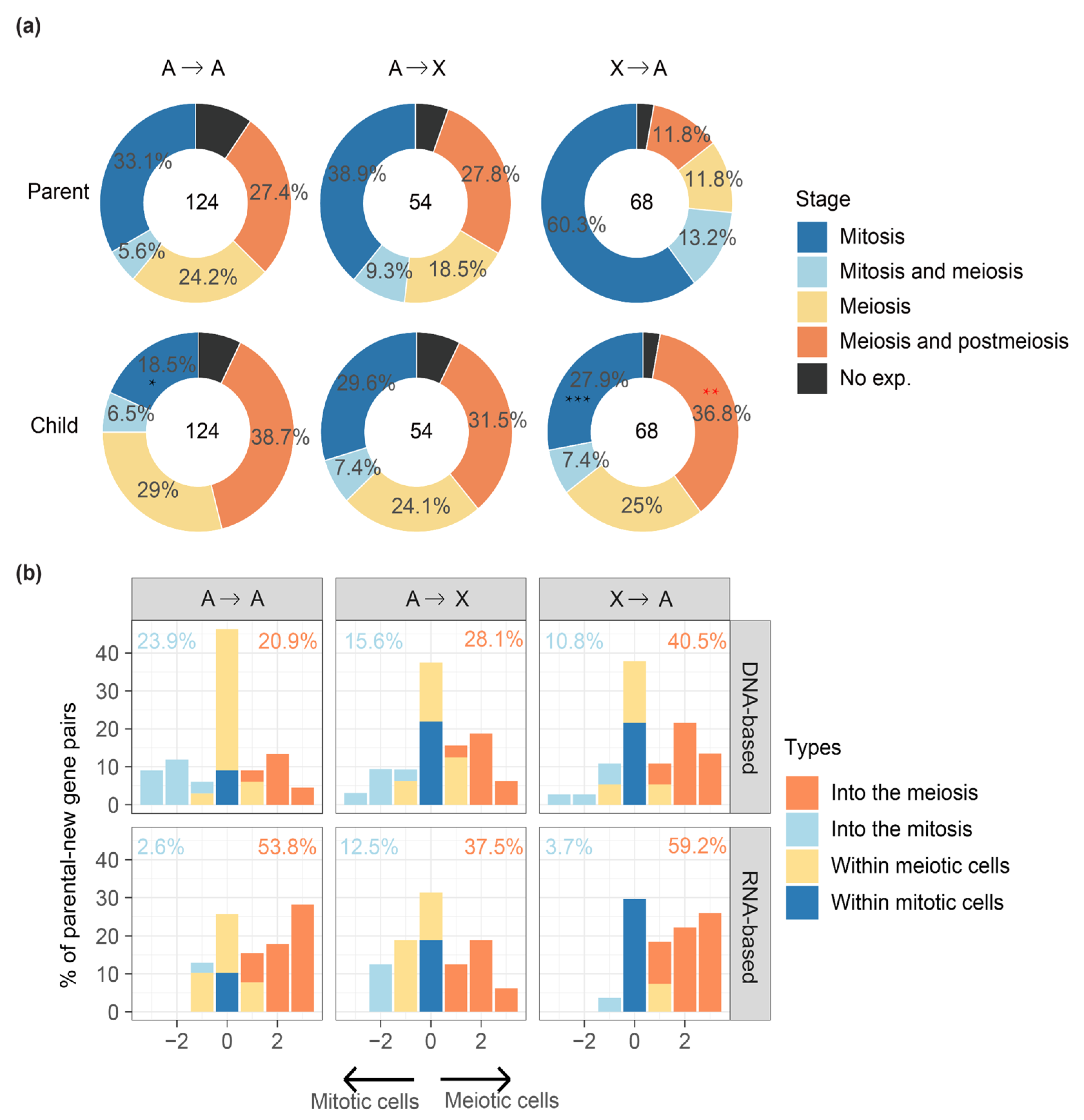
2.3. MSCI Plays an Important Role in ‘out of the X’ Pattern of New Genes
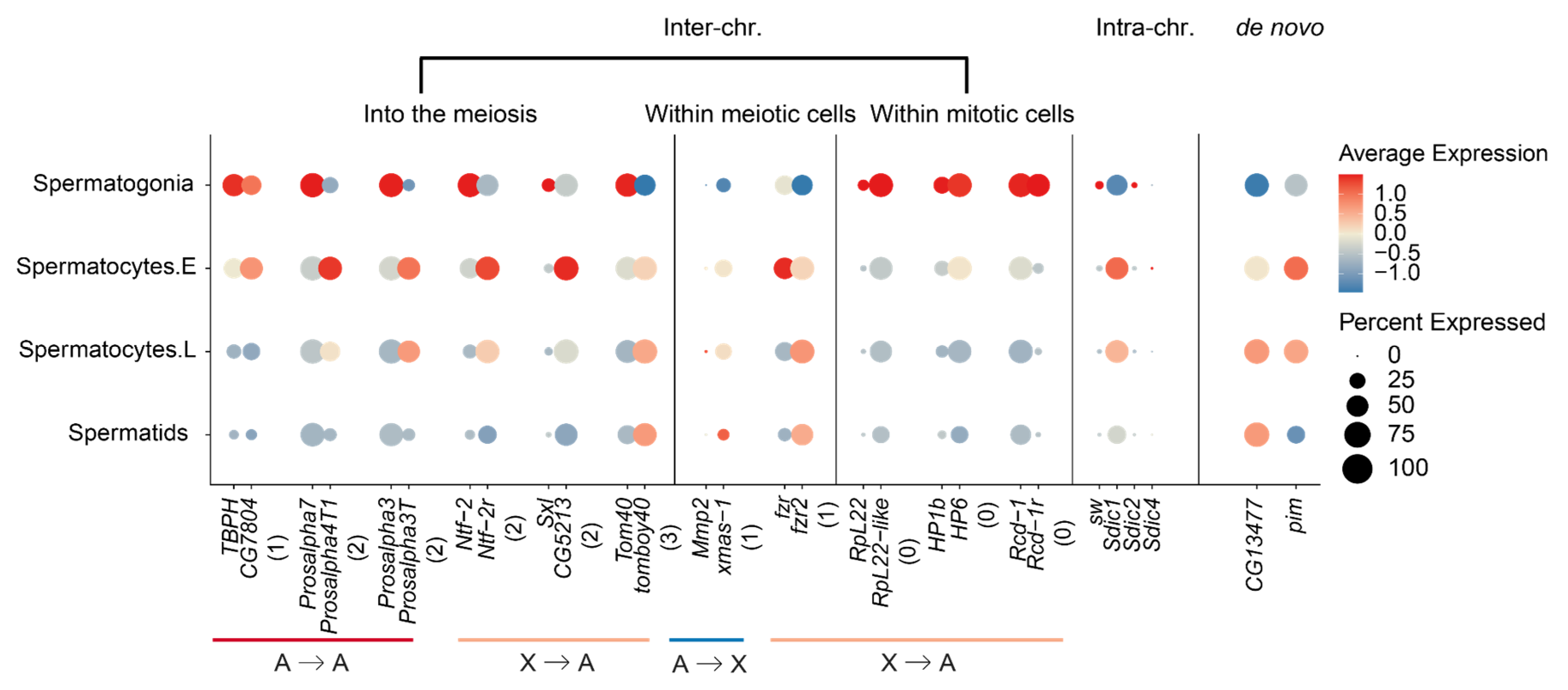
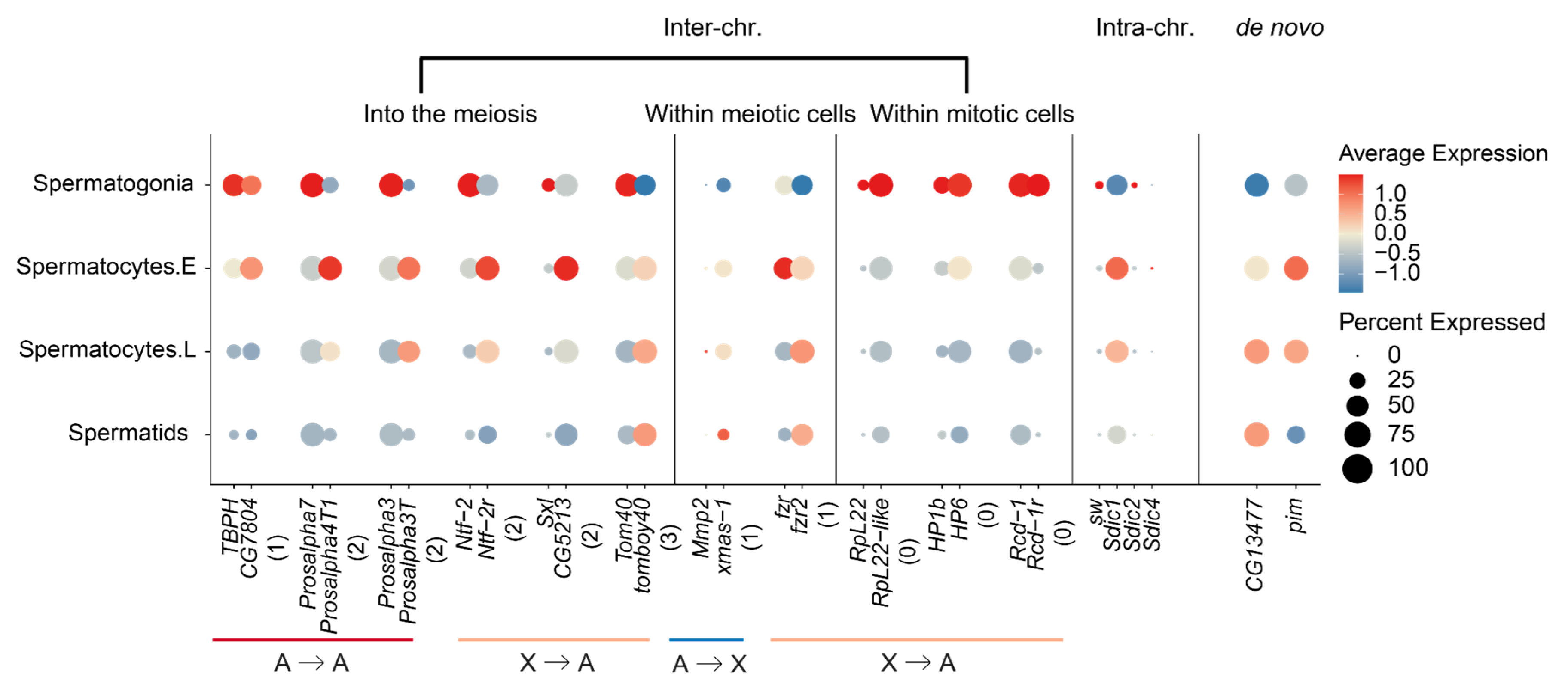
2.4. New Gene Cases of ‘into the Meiosis’
3. Discussion
4. Materials and Methods
4.1. Datasets of New Genes and scRNA-seq
4.2. Defining the Major Expressed Cell Type of Drosophila Genes
4.3. Comparing the Percentage of Genes in Different Stages between Chromosomes
4.4. Comparing the Expressed Cell Types between the New and Parental Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Long, M.; Betrán, E.; Thornton, K.; Wang, W. The origin of new genes: Glimpses from the young and old. Nat. Rev. Genet. 2003, 4, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Krinsky, B.H.; Long, M. New genes as drivers of phenotypic evolution. Nat. Rev. Genet. 2013, 14, 645–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brawand, D.; Soumillon, M.; Necsulea, A.; Julien, P.; Csárdi, G.; Harrigan, P.; Weier, M.; Liechti, A.; Aximu-Petri, A.; Kircher, M.; et al. The evolution of gene expression levels in mammalian organs. Nature 2011, 478, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Vicoso, B.; Charlesworth, B. Evolution on the X chromosome: Unusual patterns and processes. Nat. Rev. Genet. 2006, 7, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Kaessmann, H. Origins, evolution, and phenotypic impact of new genes. Genome Res. 2010, 20, 1313–1326. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.E.; Vibranovski, M.D.; Krinsky, B.H.; Long, M. Age-dependent chromosomal distribution of male-biased genes in Drosophila. Genome Res. 2010, 20, 1526–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vibranovski, M.D.; Zhang, Y.; Long, M. General gene movement off the X chromosome in the Drosophila genus. Genome Res. 2009, 19, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.-Y.; Zhou, Q. On the Regulatory Evolution of New Genes Throughout Their Life History. Mol. Biol. Evol. 2019, 36, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Kleene, K.C. A possible meiotic function of the peculiar patterns of gene expression in mammalian spermatogenic cells. Mech. Dev. 2001, 106, 3–23. [Google Scholar] [CrossRef]
- Kleene, K.C. Sexual selection, genetic conflict, selfish genes, and the atypical patterns of gene expression in spermatogenic cells. Dev. Biol. 2005, 277, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betrán, E.; Thornton, K.; Long, M. Retroposed new genes out of the X in Drosophila. Genome Res. 2002, 12, 1854–1859. [Google Scholar] [CrossRef] [Green Version]
- Emerson, J.J. Extensive Gene Traffic on the Mammalian X Chromosome. Science 2004, 303, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.M.A. Meiotic Silencing in Mammals. Annu. Rev. Genet. 2015, 49, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Potrzebowski, L.; Vinckenbosch, N.; Marques, A.C.; Chalmel, F.; Jégou, B.; Kaessmann, H. Chromosomal Gene Movements Reflect the Recent Origin and Biology of Therian Sex Chromosomes. PLoS Biol. 2008, 6, e80. [Google Scholar] [CrossRef]
- Kelly, W.G.; Schaner, C.E.; Dernburg, A.F.; Lee, M.-H.; Kim, S.K.; Villeneuve, A.M.; Reinke, V. X-chromosome silencing in the germline of C. elegans. Development 2002, 129, 479–492. [Google Scholar] [CrossRef]
- Hense, W.; Baines, J.F.; Parsch, J. X chromosome inactivation during Drosophila spermatogenesis. PLoS Biol. 2007, 5, e273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-I.; Xu, E.Y. Sexual antagonism and X inactivation—The SAXI hypothesis. Trends Genet. 2003, 19, 243–247. [Google Scholar] [CrossRef]
- Meiklejohn, C.D.; Landeen, E.L.; Cook, J.M.; Kingan, S.B.; Presgraves, D.C. Sex Chromosome-Specific Regulation in the Drosophila Male Germline But Little Evidence for Chromosomal Dosage Compensation or Meiotic Inactivation. PLoS Biol. 2011, 9, e1001126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturgill, D.; Zhang, Y.; Parisi, M.; Oliver, B. Demasculinization of X chromosomes in the Drosophila genus. Nature 2007, 450, 238–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assis, R.; Bachtrog, D. Neofunctionalization of young duplicate genes in Drosophila. Proc. Natl. Acad. Sci. USA 2013, 110, 17409–17414. [Google Scholar] [CrossRef] [Green Version]
- Vibranovski, M.D.; Lopes, H.F.; Karr, T.L.; Long, M. Stage-specific expression profiling of Drosophila spermatogenesis suggests that meiotic sex chromosome inactivation drives genomic relocation of testis-expressed genes. PLoS Genet. 2009, 5, e1000731. [Google Scholar] [CrossRef] [Green Version]
- Meiklejohn, C.D.; Presgraves, D.C. Little evidence for demasculinization of the Drosophila X chromosome among genes expressed in the male germline. Genome Biol. Evol. 2012, 4, 1007–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vibranovski, M.D.; Zhang, Y.E.; Kemkemer, C.; Lopes, H.F.; Karr, T.L.; Long, M. Re-analysis of the larval testis data on meiotic sex chromosome inactivation revealed evidence for tissue-specific gene expression related to the Drosophila X chromosome. BMC Biol. 2012, 10, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahadevaraju, S.; Fear, J.M.; Akeju, M.; Galletta, B.J.; Pinheiro, M.M.L.S.; Avelino, C.C.; Cabral-de-Mello, D.C.; Conlon, K.; Dell’Orso, S.; Demere, Z.; et al. Dynamic sex chromosome expression in Drosophila male germ cells. Nat. Commun. 2021, 12, 892. [Google Scholar] [CrossRef] [PubMed]
- Vicoso, B.; Bachtrog, D. Reversal of an ancient sex chromosome to an autosome in Drosophila. Nature 2013, 499, 332–335. [Google Scholar] [CrossRef] [Green Version]
- Witt, E.; Benjamin, S.; Svetec, N.; Zhao, L. Testis single-cell RNA-seq reveals the dynamics of de novo gene transcription and germline mutational bias in Drosophila. Elife 2019, 8, e47138. [Google Scholar] [CrossRef]
- Shao, Y.; Chen, C.; Shen, H.; He, B.Z.; Yu, D.; Jiang, S.; Zhao, S.; Gao, Z.; Zhu, Z.; Chen, X.; et al. GenTree, an integrated resource for analyzing the evolution and function of primate-specific coding genes. Genome Res. 2019, 29, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.H.; Jenkins, P.A.; Song, Y.S. Genome-wide fine-scale recombination rate variation in Drosophila melanogaster. PLoS Genet. 2012, 8, e1003090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Wang, W. On the origin and evolution of new genes—A genomic and experimental perspective. J. Genet. Genom. 2008, 35, 639–648. [Google Scholar] [CrossRef]
- Khil, P.P.; Smirnova, N.A.; Romanienko, P.J.; Camerini-Otero, R.D. The mouse X chromosome is enriched for sex-biased genes not subject to selection by meiotic sex chromosome inactivation. Nat. Genet. 2004, 36, 642–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betrán, E.; Long, M. Dntf-2r, a young Drosophila retroposed gene with specific male expression under positive Darwinian selection. Genetics 2003, 164, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Fukunaga, R. RNA-binding protein Maca is crucial for gigantic male fertility factor gene expression, spermatogenesis, and male fertility, in Drosophila. PLoS Genet. 2021, 17, e1009655. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Vedanayagam, J.; Mohammed, J.; Eizadshenass, S.; Kan, L.; Pang, N.; Aradhya, R.; Siepel, A.; Steinhauer, J.; Lai, E.C. New genes often acquire male-specific functions but rarely become essential in Drosophila. Genes Dev. 2017, 31, 1841–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page-McCaw, A.; Serano, J.; Santé, J.M.; Rubin, G.M. Drosophila Matrix Metalloproteinases Are Required for Tissue Remodeling, but Not Embryonic Development. Dev. Cell 2003, 4, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Deady, L.D.; Shen, W.; Mosure, S.A.; Spradling, A.C.; Sun, J. Matrix metalloproteinase 2 is required for ovulation and corpus luteum formation in Drosophila. PLoS Genet. 2015, 11, e1004989. [Google Scholar] [CrossRef] [Green Version]
- Jevitt, A.; Chatterjee, D.; Xie, G.; Wang, X.-F.; Otwell, T.; Huang, Y.-C.; Deng, W.-M. A single-cell atlas of adult Drosophila ovary identifies transcriptional programs and somatic cell lineage regulating oogenesis. PLoS Biol. 2020, 18, e3000538. [Google Scholar] [CrossRef] [PubMed]
- McKim, K.S.; Dahmus, J.B.; Hawley, R.S. Cloning of the Drosophila melanogaster meiotic recombination gene mei-218: A genetic and molecular analysis of interval 15E. Genetics 1996, 144, 215–228. [Google Scholar] [CrossRef]
- Gallach, M.; Betrán, E. Intralocus sexual conflict resolved through gene duplication. Trends Ecol. Evol. 2011, 26, 222–228. [Google Scholar] [CrossRef] [Green Version]
- VanKuren, N.W.; Long, M. Gene duplicates resolving sexual conflict rapidly evolved essential gametogenesis functions. Nat. Ecol. Evol. 2018, 2, 705–712. [Google Scholar] [CrossRef]
- Yeh, S.-D.; Do, T.; Abbassi, M.; Ranz, J.M. Functional relevance of the newly evolved sperm dynein intermediate chain multigene family in Drosophila melanogaster males. Commun. Integr. Biol. 2012, 5, 462–465. [Google Scholar] [CrossRef]
- Conrad, T.; Akhtar, A. Dosage compensation in Drosophila melanogaster: Epigenetic fine-tuning of chromosome-wide transcription. Nat. Rev. Genet. 2012, 13, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Witt, E.; Shao, Z.; Hu, C.; Krause, H.M.; Zhao, L. Single-cell RNA-sequencing reveals pre-meiotic X-chromosome dosage compensation in Drosophila testis. PLoS Genet. 2021, 17, e1009728. [Google Scholar] [CrossRef] [PubMed]
- Gan, Q.; Schones, D.E.; Ho Eun, S.; Wei, G.; Cui, K.; Zhao, K.; Chen, X. Monovalent and unpoised status of most genes in undifferentiated cell-enriched Drosophila testis. Genome Biol. 2010, 11, R42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, Q.; Chepelev, I.; Wei, G.; Tarayrah, L.; Cui, K.; Zhao, K.; Chen, X. Dynamic regulation of alternative splicing and chromatin structure in Drosophila gonads revealed by RNA-seq. Cell Res. 2010, 20, 763–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.J.; McCarrey, J.R.; Yang, F.; Page, D.C. An abundance of X-linked genes expressed in spermatogonia. Nat. Genet. 2001, 27, 422–426. [Google Scholar] [CrossRef]
- Rice, W.R. Sex Chromosomes and the Evolution of Sexual Dimorphism. Evolution 1984, 38, 735. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, B.; Coyne, J.A.; Barton, N.H. The Relative Rates of Evolution of Sex Chromosomes and Autosomes. Am. Nat. 1987, 130, 113–146. [Google Scholar] [CrossRef]
- Haniffa, M.; Taylor, D.; Linnarsson, S.; Aronow, B.J.; Bader, G.D.; Barker, R.A.; Camara, P.G.; Camp, J.G.; Chédotal, A.; Copp, A.; et al. A roadmap for the Human Developmental Cell Atlas. Nature 2021, 597, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [Green Version]
- McGinnis, C.S.; Murrow, L.M.; Gartner, Z.J. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 2019, 8, 329–337.e324. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- Hafemeister, C.; Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019, 20, 296. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graveley, B. The developmental transcriptome of Drosophila melanogaster. Genome Biol. 2010, 11, 473–479. [Google Scholar] [CrossRef] [Green Version]
- Daines, B.; Wang, H.; Wang, L.; Li, Y.; Han, Y.; Emmert, D.; Gelbart, W.; Wang, X.; Li, W.; Gibbs, R.; et al. The Drosophila melanogaster transcriptome by paired-end RNA sequencing. Genome Res. 2011, 21, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, Q.; He, H.; Zhou, Q. On the Origin and Evolution of Drosophila New Genes during Spermatogenesis. Genes 2021, 12, 1796. https://doi.org/10.3390/genes12111796
Su Q, He H, Zhou Q. On the Origin and Evolution of Drosophila New Genes during Spermatogenesis. Genes. 2021; 12(11):1796. https://doi.org/10.3390/genes12111796
Chicago/Turabian StyleSu, Qianwei, Huangyi He, and Qi Zhou. 2021. "On the Origin and Evolution of Drosophila New Genes during Spermatogenesis" Genes 12, no. 11: 1796. https://doi.org/10.3390/genes12111796
APA StyleSu, Q., He, H., & Zhou, Q. (2021). On the Origin and Evolution of Drosophila New Genes during Spermatogenesis. Genes, 12(11), 1796. https://doi.org/10.3390/genes12111796