
Genetically Targeted Clinical Trials in Parkinson’s Disease: Learning from the Successes Made in Oncology


{kind=link}
{kind=link}
Abstract
:1. Introduction
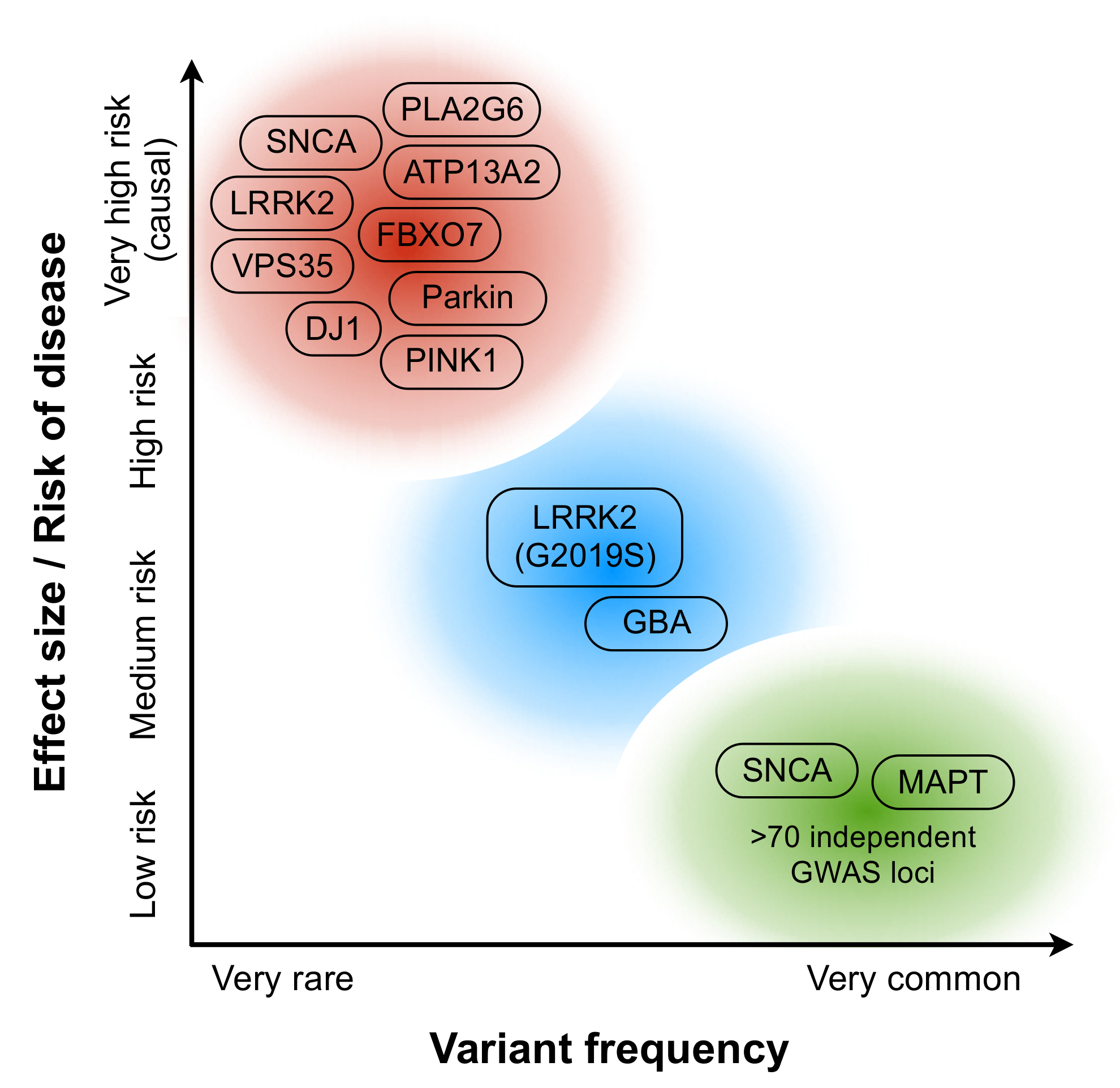
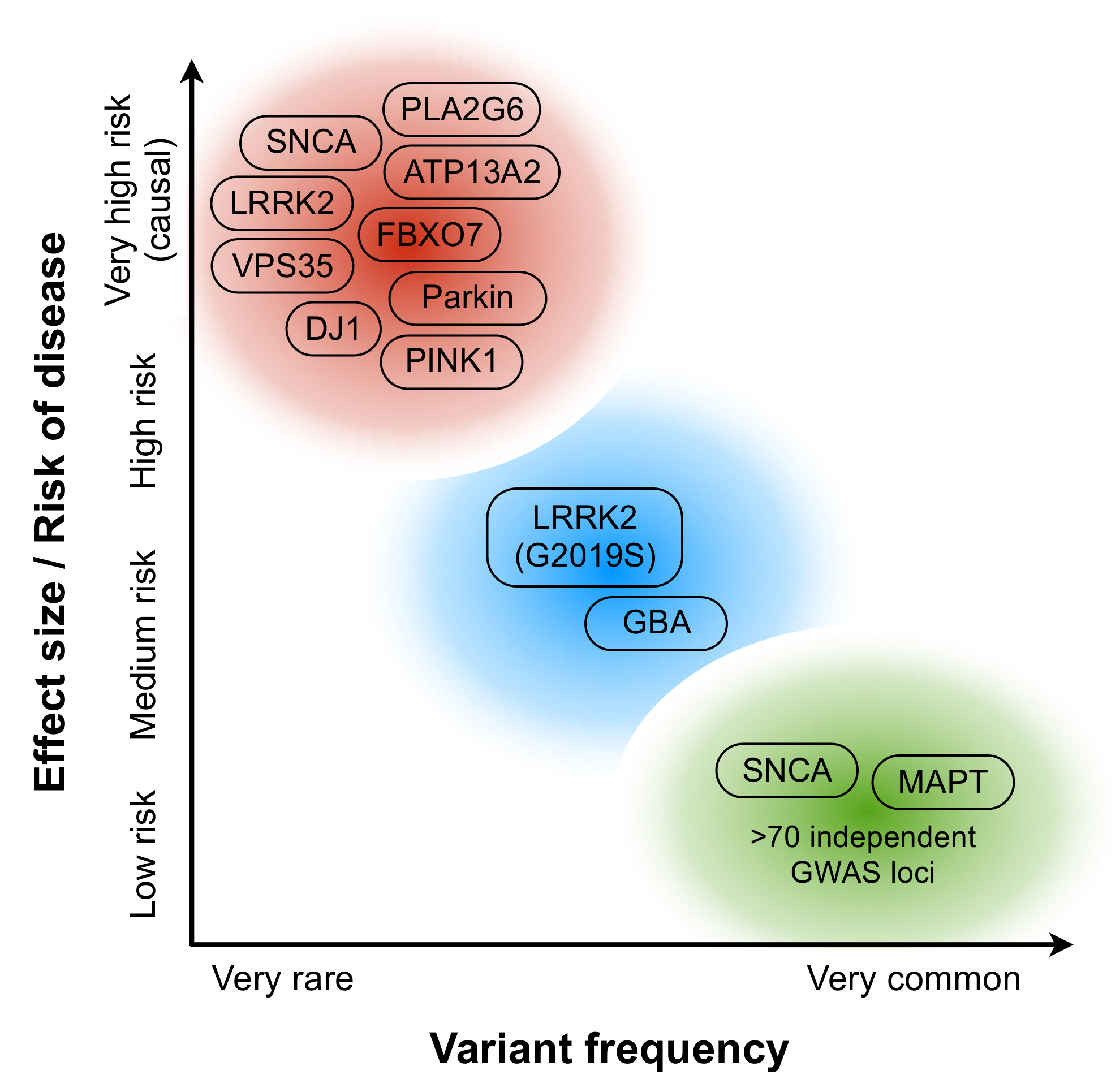
2. Disease Biology in PD
3. Bringing Knowledge of Disease Biology into Clinical Trials
4. Challenges and Opportunities of Genetically Targeted Clinical Trials in PD
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Wexler, M. Biogen Discontinues Development of Cinpanemab. Available online: https://parkinsonsnewstoday.com/2021/02/04/biogen-announcement-discontinue-cinpanemab-parkinsons/ (accessed on 4 February 2021).
- FDA. Hematology/Oncology (Cancer) Approvals & Safety Notifications. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/hematologyoncology-cancer-approvals-safety-notifications (accessed on 28 May 2021).
- Rose, E. Update on Phase 2 Pasadena Study of Prasinezumab (Prx002/Rg7935) in Parkinson’s Disease. Available online: https://ir.prothena.com/news-releases/news-release-details/update-phase-2-pasadena-study-prasinezumab-prx002rg7935 (accessed on 30 May 2021).
- Munoz, E.; Oliva, R.; Obach, V.; Marti, M.J.; Pastor, P.; Ballesta, F.; Tolosa, E. Identification of Spanish familial Parkinson’s disease and screening for the Ala53Thr mutation of the alpha-synuclein gene in early onset patients. Neurosci. Lett. 1997, 235, 57–60. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Scott, W.K.; Staijich, J.M.; Yamaoka, L.H.; Speer, M.C.; Vance, J.M.; Roses, A.D.; Pericak-Vance, M.A. Genetic complexity and Parkinson’s disease. Deane Laboratory Parkinson Disease Research Group. Science 1997, 277, 387–388. [Google Scholar] [CrossRef] [Green Version]
- Shulman, J.M.; De Jager, P.L.; Feany, M.B. Parkinson’s Disease: Genetics and Pathogenesis. Annu. Rev. Pathol. 2011, 6, 193–222. [Google Scholar] [CrossRef] [Green Version]
- Adler, C.H. Differential diagnosis of Parkinson’s disease. Med. Clin. N. Am. 1999, 83, 349–367. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, T.P.; Rumsby, P.C.; Capleton, A.C.; Rushton, L.; Levy, L.S. Pesticides and Parkinson’s Disease—Is There a Link? Environ. Health Perspect. 2006, 114, 156–164. [Google Scholar] [CrossRef]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Levy, G. The Relationship of Parkinson Disease with Aging. Arch. Neurol. 2007, 64, 1242–1246. [Google Scholar] [CrossRef] [PubMed]
- Tran, J.; Anastacio, H.; Bardy, C. Genetic predispositions of Parkinson’s disease revealed in patient-derived brain cells. NPJ Parkinsons Dis. 2020, 6, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherian, A.; Divya, K.P. Genetics of Parkinson’s disease. Acta Neurol. Belg. 2020, 120, 1297–1305. [Google Scholar] [CrossRef]
- Lázaro, D.F.; Outeiro, T.F. The Interplay Between Proteostasis Systems and Parkinson’s Disease. Adv. Exp. Med. Biol. 2020, 1233, 223–236. [Google Scholar] [CrossRef]
- Li, W.; Fu, Y.; Halliday, G.M.; Sue, C.M. PARK Genes Link Mitochondrial Dysfunction and Alpha-Synuclein Pathology in Sporadic Parkinson’s Disease. Front. Cell Dev. Biol. 2021, 9, 612476. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Kaur, G.; Fratila, O.; Buhas, C.; Judea-Pusta, C.T.; Negrut, N.; Bustea, C.; Bungau, S. Cross-talks among GBA mutations, glucocerebrosidase, and α-synuclein in GBA-associated Parkinson’s disease and their targeted therapeutic approaches: A comprehensive review. Transl. Neurodegener. 2021, 10, 4. [Google Scholar] [CrossRef]
- Chung, E.; Choi, Y.; Park, J.; Nah, W.; Park, J.; Jung, Y.; Lee, J.; Lee, H.; Park, S.; Hwang, S.; et al. Intracellular delivery of Parkin rescues neurons from accumulation of damaged mitochondria and pathological α-synuclein. Sci. Adv. 2020, 6, eaba1193. [Google Scholar] [CrossRef]
- Oliveras-Salvá, M.; Macchi, F.; Coessens, V.; Deleersnijder, A.; Gérard, M.; Van der Perren, A.; Haute, C.V.D.; Baekelandt, V. Alpha-synuclein-induced neurodegeneration is exacerbated in PINK1 knockout mice. Neurobiol. Aging 2014, 35, 2625–2636. [Google Scholar] [CrossRef]
- Erb, M.L.; Moore, D.J. LRRK2 and the Endolysosomal System in Parkinson’s Disease. J. Parkinsons Dis. 2020, 10, 1271–1291. [Google Scholar] [CrossRef]
- Cabezudo, D.; Baekelandt, V.; Lobbestael, E. Multiple-Hit Hypothesis in Parkinson’s Disease: LRRK2 and Inflammation. Front. Neurosci. 2020, 14, 376. [Google Scholar] [CrossRef] [PubMed]
- Fanning, S.; Selkoe, D.; Dettmer, U. Parkinson’s disease: Proteinopathy or lipidopathy? NPJ Parkinsons Dis. 2020, 6, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paslawski, W.; Zareba-Paslawska, J.; Zhang, X.; Hölzl, K.; Wadensten, H.; Shariatgorji, R.; Janelidze, S.; Hansson, O.; Forsgren, L.; Andrén, P.E.; et al. α-synuclein−lipoprotein interactions and elevated ApoE level in cerebrospinal fluid from Parkinson’s disease patients. Proc. Natl. Acad. Sci. USA 2019, 116, 15226–15235. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.E.; Stecher, B.; Labrie, V.; Brundin, L.; Brundin, P. Triggers, Facilitators, and Aggravators: Redefining Parkinson’s Disease Pathogenesis. Trends Neurosci. 2019, 42, 4–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holden, S.; Finseth, T.; Sillau, S.H.; Berman, B.D. Progression of MDS-UPDRS Scores Over Five Years in De Novo Parkinson Disease from the Parkinson’s Progression Markers Initiative Cohort. Mov. Disord. Clin. Pract. 2018, 5, 47–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venuto, C.S.; Potter, N.B.; Dorsey, E.R.; Kieburtz, K. A review of disease progression models of Parkinson’s disease and applications in clinical trials. Mov. Disord. 2016, 31, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Mullin, S.; Smith, L.; Lee, K.; D’Souza, G.; Woodgate, P.; Elflein, J.; Hällqvist, J.; Toffoli, M.; Streeter, A.; Hosking, J.; et al. Ambroxol for the Treatment of Patients with Parkinson Disease with and without Glucocerebrosidase Gene Mutations: A Nonrandomized, Noncontrolled Trial. JAMA Neurol. 2020, 77, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Collins, L.M.; Murphy, S.A.; Strecher, V. The Multiphase Optimization Strategy (MOST) and the Sequential Multiple Assignment Randomized Trial (SMART): New Methods for More Potent eHealth Interventions. Am. J. Prev. Med. 2007, 32, S112–S118. [Google Scholar] [CrossRef] [Green Version]
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Bandrés-Ciga, S.; Ruz, C.; Barrero, F.J.; Escamilla-Sevilla, F.; Pelegrina, J.; Vives, F.; Duran, R. Structural genomic variations and Parkinson’s disease. Minerva Med. 2017, 108, 438–447. [Google Scholar] [CrossRef]
- Saunders-Pullman, R.; Lipton, R.B.; Senthil, G.; Katz, M.; Costan-Toth, C.; Derby, C.; Bressman, S.; Verghese, J.; Ozelius, L.J. Increased frequency of the LRRK2 G2019S mutation in an elderly Ashkenazi Jewish population is not associated with dementia. Neurosci. Lett. 2006, 402, 92–96. [Google Scholar] [CrossRef]
- Benamer, H.T.; De Silva, R. LRRK2 G2019S in the North African Population: A Review. Eur. Neurol. 2010, 63, 321–325. [Google Scholar] [CrossRef]
- Sidransky, E.; Lopez, G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012, 11, 986–998. [Google Scholar] [CrossRef] [Green Version]
- Koval, I.; Bône, A.; Louis, M.; Lartigue, T.; Bottani, S.; Marcoux, A.; Samper-González, J.; Burgos, N.; Charlier, B.; Bertrand, A.; et al. AD Course Map charts Alzheimer’s disease progression. Sci. Rep. 2021, 11, 8020. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sjögren, M.; Huttunen, H.J.; Svenningsson, P.; Widner, H. Genetically Targeted Clinical Trials in Parkinson’s Disease: Learning from the Successes Made in Oncology. Genes 2021, 12, 1529. https://doi.org/10.3390/genes12101529
Sjögren M, Huttunen HJ, Svenningsson P, Widner H. Genetically Targeted Clinical Trials in Parkinson’s Disease: Learning from the Successes Made in Oncology. Genes. 2021; 12(10):1529. https://doi.org/10.3390/genes12101529
Chicago/Turabian StyleSjögren, Magnus, Henri J. Huttunen, Per Svenningsson, and Håkan Widner. 2021. "Genetically Targeted Clinical Trials in Parkinson’s Disease: Learning from the Successes Made in Oncology" Genes 12, no. 10: 1529. https://doi.org/10.3390/genes12101529
APA StyleSjögren, M., Huttunen, H. J., Svenningsson, P., & Widner, H. (2021). Genetically Targeted Clinical Trials in Parkinson’s Disease: Learning from the Successes Made in Oncology. Genes, 12(10), 1529. https://doi.org/10.3390/genes12101529