
Roles for α-Synuclein in Gene Expression

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Molecular Organization of α-Syn and Its Role in Pathology
1.1. Synucleinopathies
1.2. Therapeutic Silencing of α-Syn
2. Role of α-Syn in Regulating Gene Expression
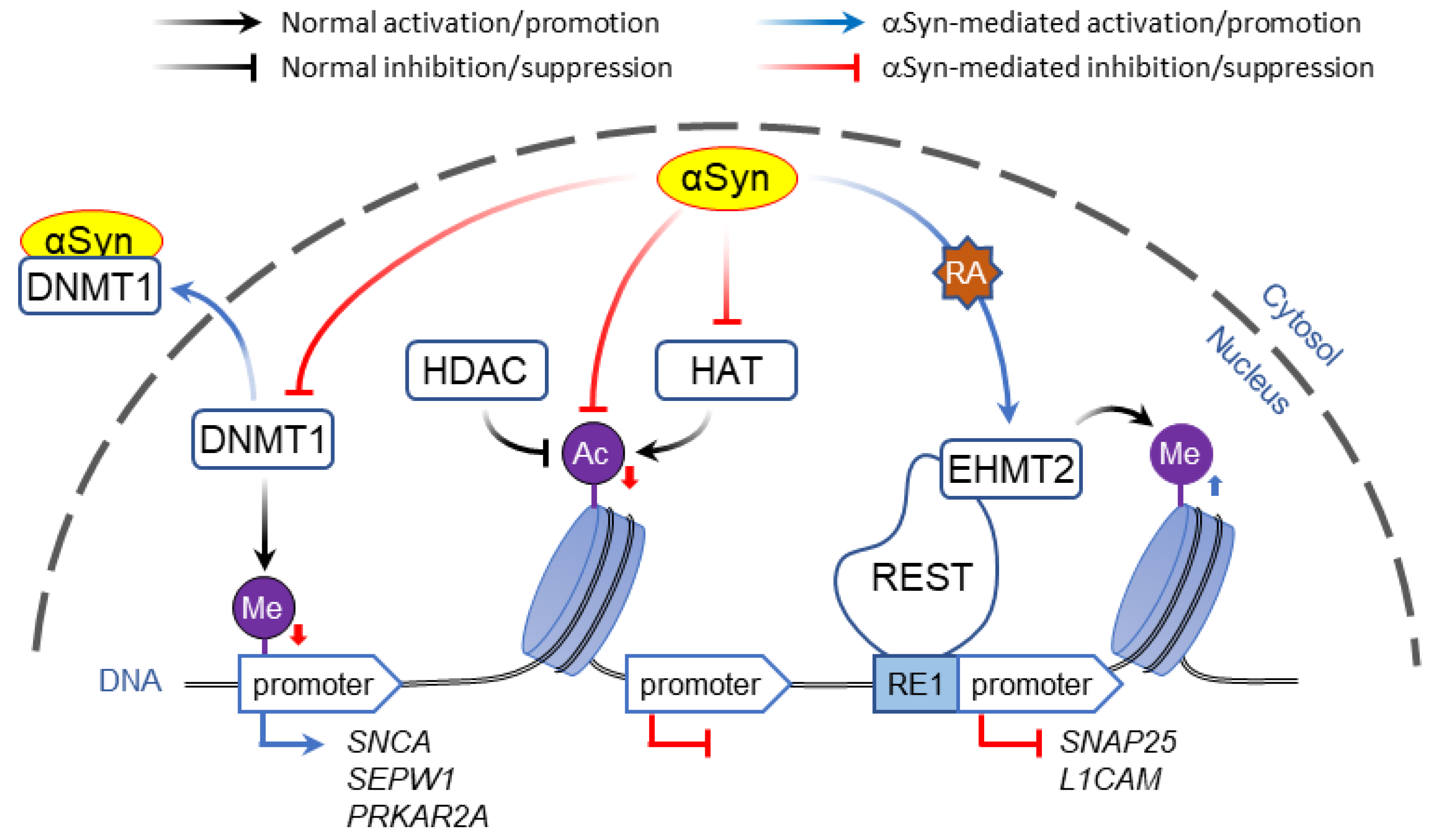
2.1. Interaction of α-Syn with DNA and Histones
2.1.1. Direct DNA Binding
2.1.2. Interaction with Histones
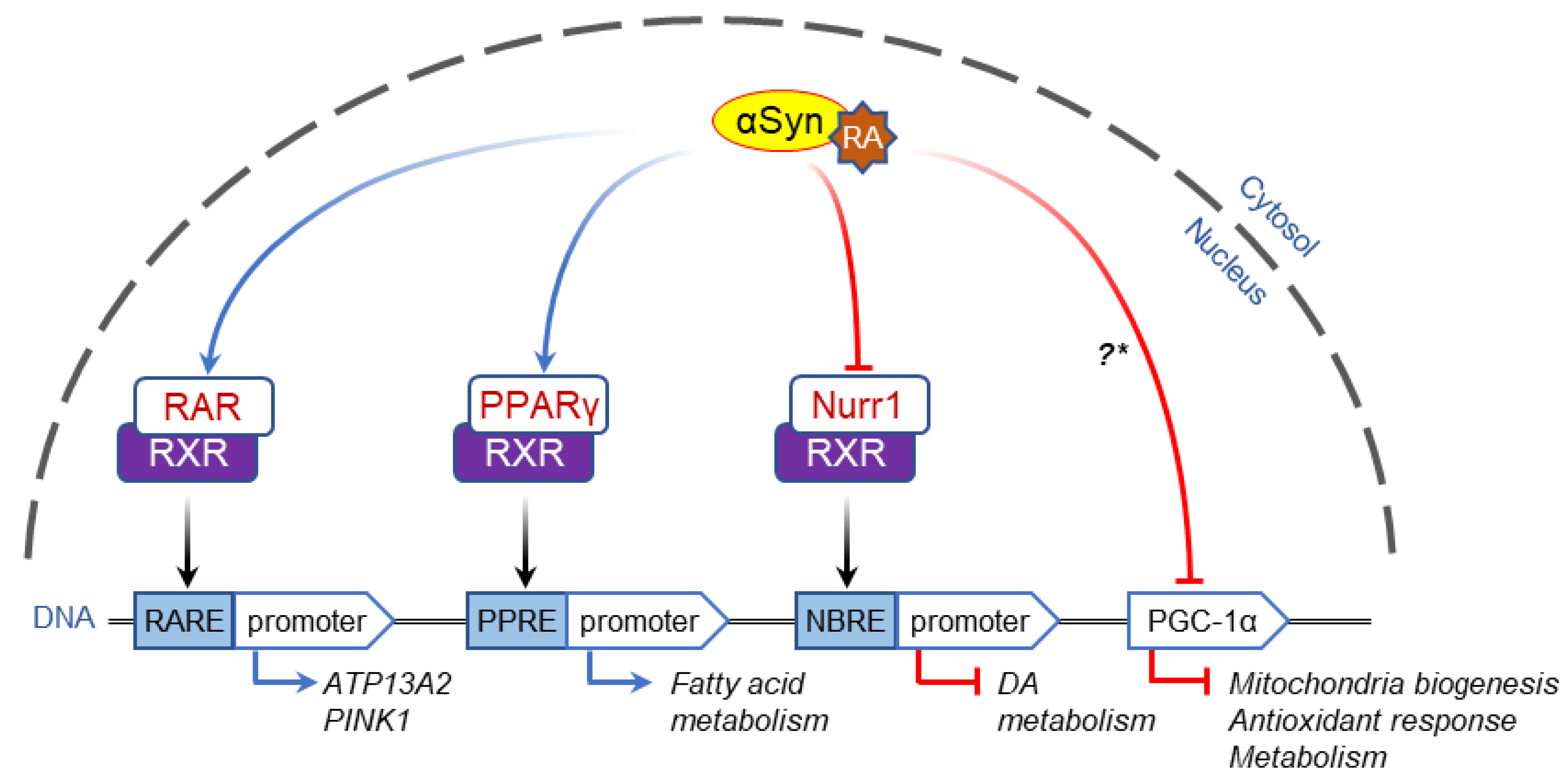
2.2. α-Syn-Dependent Regulation of Nuclear Receptors
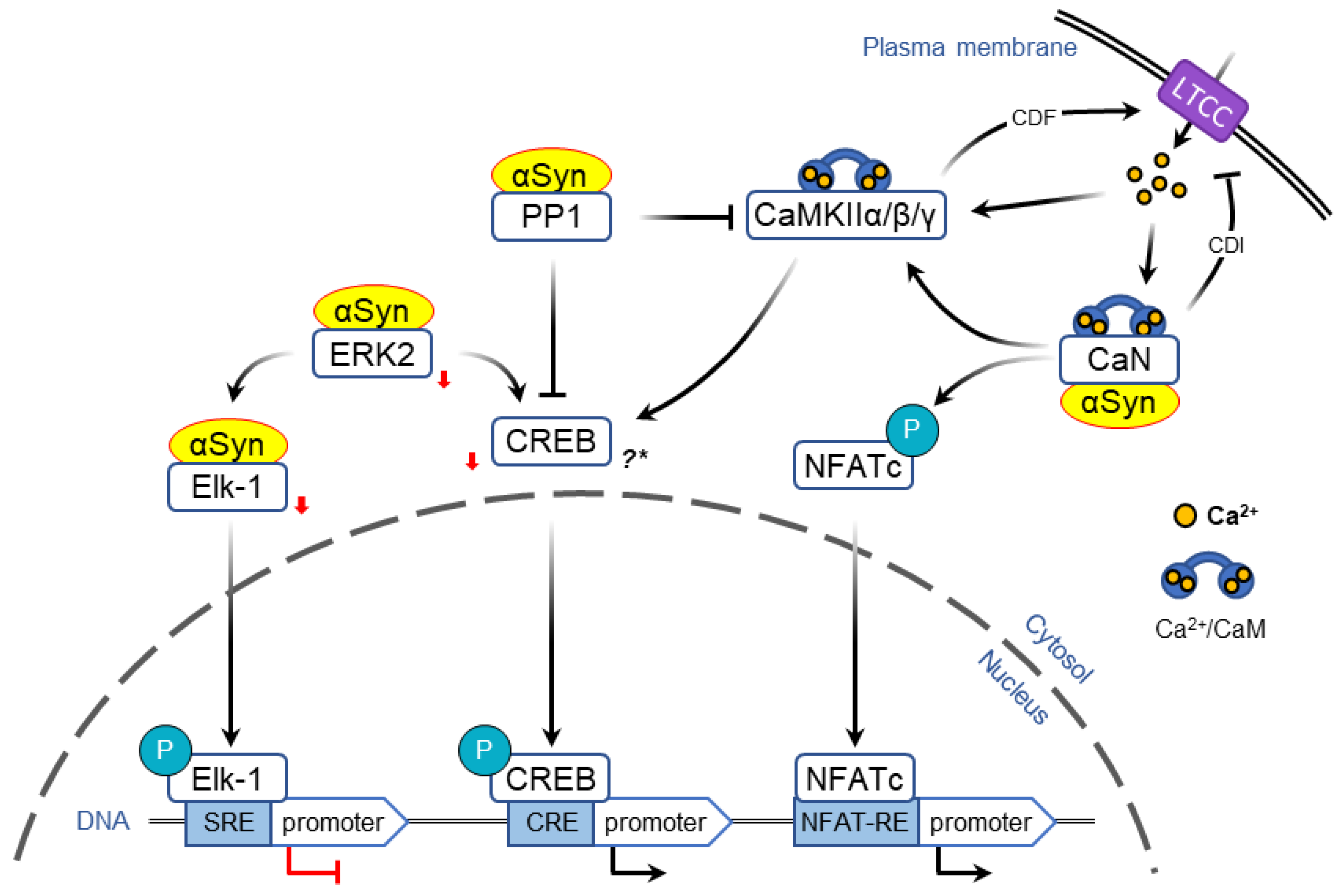
2.3. α-Syn-Dependent Regulation of Immediate Early Genes
2.3.1. Regulation of Transcription via MAPK/ERK Pathway
2.3.2. Regulation of Transcription via LTCC/CaMK Pathway
3. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AADC | Aromatic L-amino acid decarboxylase |
| CaN | calcineurin |
| CaM | Calmodulin |
| CaMK | CaM-dependent protein kinases |
| CRE | cAMP response element |
| pCREB | phospho-cyclic-AMP-response-element-binding protein |
| DA | dopamine |
| DNMT1 | DNA methyl transferases 1 |
| EF sequence | Elongation Factor sequence |
| EHMT2 | euchromatic histone lysine N-methyltransferase 2 |
| Elk-1 | ETS Like-1 protein |
| ERK2 | Extracellular signal-regulated kinase 2 (a.k.a. MAPK1: mitogen-activated protein kinase 1) |
| GCH 1 | GTP Cyclohydrolase 1 |
| HAT | histone acetyltransferase |
| HDAC | histone deacetylase |
| LTCC | L-type calcium channels |
| MAPK | Mitogen activated protein kinase |
| NBRE | NGFI-B response element |
| NFATc/n | Nuclear and cytosolic factors of activated T cells |
| NFAT-RE | nuclear factor of activated T-cells response element |
| Nurr1 | nuclear receptor related 1 |
| PD | Parkinson’s Disease |
| PTM | post-translational modification |
| PPARγ | peroxisome proliferator-activated receptor-γ |
| PGC-1α | receptor γ coactivator-1 |
| RA | retinoic acid |
| RAR | Retinoic-acid receptor |
| RARE | retinoic acid response element |
| REST | repressor element-1 silencing transcription factor (a.k.a. NRSF: neuron-restrictive silencer factor) |
| RXR | retinoid-X receptors |
| α-Syn | α-Synuclein |
| SRE | Serum response element |
| TH | Tyrosine Hydroxylase |
References
- Bartels, T.; Choi, J.G.; Selkoe, D.J. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Jakes, R.; Spillantini, M.G.; Goedert, M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994, 345, 27–32. [Google Scholar] [CrossRef]
- Dettmer, U.; Newman, A.J.; von Saucken, V.; Bartels, T.; Selkoe, D. KTKEGV repeat motifs are key mediators of normal alpha-synuclein tetramerization: Their mutation causes excess monomers and neurotoxicity. Proc. Natl. Acad. Sci. USA 2015, 112, 9596–9601. [Google Scholar] [CrossRef]
- Terakawa, M.S.; Lin, Y.; Kinoshita, M.; Kanemura, S.; Itoh, D.; Sugiki, T.; Okumura, M.; Ramamoorthy, A.; Lee, Y.-H. Impact of membrane curvature on amyloid aggregation. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1741–1764. [Google Scholar] [CrossRef] [PubMed]
- Segrest, J.P.; Jones, M.K.; De Loof, H.; Brouillette, C.G.; Venkatachalapathi, Y.V.; Anantharamaiah, G.M. The amphipathic helix in the exchangeable apolipoproteins: A review of secondary structure and function. J. Lipid Res. 1992, 33, 141–166. [Google Scholar] [CrossRef]
- Sulzer, D.; Edwards, R.H. The physiological role of alpha-synuclein and its relationship to Parkinson’s Disease. J. Neurochem. 2019, 150, 475–486. [Google Scholar] [CrossRef]
- Lautenschläger, J.; Kaminski, C.F.; Schierle, G.S.K. alpha-Synuclein—Regulator of Exocytosis, Endocytosis, or Both? Trends Cell Biol. 2017, 27, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Li, H.T.; Du, H.N.; Tang, L.; Hu, J.; Hu, H.Y. Structural transformation and aggregation of human alpha-synuclein in trifluoroethanol: Non-amyloid component sequence is essential and beta-sheet formation is prerequisite to aggregation. Biopolymers 2002, 64, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, I.J.; Pervaiz, N.; Abbasi, A.A. The Parkinson Disease gene SNCA: Evolutionary and structural insights with pathological implication. Sci. Rep. 2016, 6, 24475. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, S.; Ogaki, K.; Tacik, P.M.; Uitti, R.J.; Ross, O.A.; Wszolek, Z.K. Update on novel familial forms of Parkinson’s disease and multiple system atrophy. Parkinsonism Relat. Disord. 2014, 20, S29–S34. [Google Scholar] [CrossRef]
- Stefanis, L. alpha-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Armakola, M.; Dumoulin, M.; Parastatidis, I.; Ischiropoulos, H. Cellular Oligomerization of alpha-Synuclein Is Determined by the Interaction of Oxidized Catechols with a C-terminal Sequence. J. Biol. Chem. 2007, 282, 31621–31630. [Google Scholar] [CrossRef]
- Herrera, F.E.; Chesi, A.; Paleologou, K.E.; Schmid, A.; Munoz, A.; Vendruscolo, M.; Gustincich, S.; Lashuel, H.A.; Carloni, P. Inhibition of alpha-Synuclein Fibrillization by Dopamine Is Mediated by Interactions with Five C-Terminal Residues and with E83 in the NAC Region. PLoS ONE 2008, 3, e003394. [Google Scholar] [CrossRef]
- Conway, K.A.; Rochet, J.-C.; Bieganski, R.M.; Lansbury, P.T. Kinetic Stabilization of the alpha -Synuclein Protofibril by a Dopamine-alpha-Synuclein Adduct. Science 2001, 294, 1346–1349. [Google Scholar] [CrossRef]
- Rochet, J.C.; Outeiro, T.F.; Conway, K.A.; Ding, T.T.; Volles, M.J.; Lashuel, H.A.; Bieganski, R.M.; Lindquist, S.L.; Lansbury, P.T. Interactions among alpha-synuclein, dopamine, and biomembranes: Some clues for understanding neurodegeneration in Parkinson’s disease. J. Mol. Neurosci. 2004, 23, 23–34. [Google Scholar] [CrossRef]
- Lautenschläger, J.; Stephens, A.D.; Fusco, G.; Stroehl, F.; Curry, N.; Zacharopoulou, M.; Michel, C.H.; Laine, R.; Nespovitaya, N.; Fantham, M.; et al. C-terminal calcium binding of alpha-synuclein modulates synaptic vesicle interaction. Nat. Commun. 2018, 9, 712. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.S.; Vorum, H.; Lindersson, E.; Jensen, P.H. Ca2+ Binding to alpha-Synuclein Regulates Ligand Binding and Oligomerization. J. Biol. Chem. 2001, 276, 22680–22684. [Google Scholar] [CrossRef]
- Tofaris, G.K.; Reitböck, P.G.; Humby, T.; Lambourne, S.L.; O’Connell, M.; Ghetti, B.; Gossage, H.; Emson, P.C.; Wilkinson, L.S.; Goedert, M.; et al. Pathological Changes in Dopaminergic Nerve Cells of the Substantia Nigra and Olfactory Bulb in Mice Transgenic for Truncated Human alpha-Synuclein(1–120): Implications for Lewy Body Disorders. J. Neurosci. 2006, 26, 3942–3950. [Google Scholar] [CrossRef] [PubMed]
- Oueslati, A. Implication of Alpha-Synuclein Phosphorylation at S129 in Synucleinopathies: What Have We Learned in the Last Decade? J. Park. Dis. 2016, 6, 39–51. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M. Neurodegeneration and the ordered assembly of alpha-synuclein. Cell Tissue Res. 2018, 373, 137–148. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A.; Lantos, P.L. Papp-Lantos inclusions and the pathogenesis of multiple system atrophy: An update. Acta Neuropathol. 2010, 119, 657–667. [Google Scholar] [CrossRef]
- Marras, C.; Beck, J.C.; Bower, J.H.; Roberts, E.; Ritz, B.; Ross, G.W.; Abbott, R.D.; Savica, R.; Eeden, S.K.V.D.; Willis, A.W.; et al. Prevalence of Parkinson’s disease across North America. NPJ Park. Dis. 2018, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W. Neuropathology of Parkinson disease. Parkinsonism. Relat. Disord. 2018, 46, S30–S33. [Google Scholar] [CrossRef]
- Eriksen, J.L.; Wszolek, Z.; Petrucelli, L. Molecular Pathogenesis of Parkinson Disease. Arch. Neurol. 2005, 62, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; Van Der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef]
- Simon-Sanchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Klivenyi, P.; Siwek, D.; Gardian, G.; Yang, L.; Starkov, A.; Cleren, C.; Ferrante, R.J.; Kowall, N.W.; Abeliovich, A.; Beal, M.F. Mice lacking alpha-synuclein are resistant to mitochondrial toxins. Neurobiol. Dis. 2006, 21, 541–548. [Google Scholar] [CrossRef]
- Dauer, W.; Kholodilov, N.; Vila, M.; Trillat, A.-C.; Goodchild, R.; Larsen, K.E.; Staal, R.; Tieu, K.; Schmitz, Y.; Yuan, C.A.; et al. Resistance of alpha-synuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. USA 2002, 99, 14524–14529. [Google Scholar] [CrossRef]
- Alvarez-Fischer, D.; Henze, C.; Strenzke, C.; Westrich, J.; Ferger, B.; Höglinger, G.U.; Oertel, W.H.; Hartmann, A. Characterization of the striatal 6-OHDA model of Parkinson’s disease in wild type and alpha-synuclein-deleted mice. Exp. Neurol. 2008, 210, 182–193. [Google Scholar] [CrossRef]
- Jensen, P.H.; Hager, H.; Nielsen, M.S.; Højrup, P.; Gliemann, J.; Jakes, R. α-Synuclein Binds to Tau and Stimulates the Protein Kinase A-catalyzed Tau Phosphorylation of Serine Residues 262 and 356. J. Biol. Chem. 1999, 274, 25481–25489. [Google Scholar] [CrossRef]
- Feng, S.T.; Wang, Z.Z.; Yuan, Y.H.; Sun, H.M.; Chen, N.H.; Zhang, Y. Update on the association between alpha-synuclein and tau with mitochondrial dysfunction: Implications for Parkinson’s disease. Eur. J. Neurosci. 2020, 59, 2946–2959. [Google Scholar] [CrossRef] [PubMed]
- Daher, J.P. Interaction of LRRK2 and alpha-Synuclein in Parkinson’s Disease. Adv. Neurobiol. 2017, 14, 209–226. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Yamashita, H.; Takahashi, T.; Nakamura, S. Activated Fyn Phosphorylates alpha-Synuclein at Tyrosine Residue 125. Biochem. Biophys. Res. Commun. 2001, 280, 1085–1092. [Google Scholar] [CrossRef]
- Choi, P.; Golts, N.; Snyder, H.; Chong, M.; Petrucelli, L.; Hardy, J.; Sparkman, D.; Cochran, E.; Lee, J.M.; Wolozin, B. Co-association of parkin and alpha-synuclein. NeuroReport 2001, 12, 2839–2843. [Google Scholar] [CrossRef]
- Zhu, M.; Patel, S.H.; Han, S. DJ-1, a Parkinson’s disease related protein, aggregates under denaturing conditions and co-aggregates with alpha-synuclein through hydrophobic interaction. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1759–1769. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E.; Hazrati, L.-N.; Fujioka, S.; Wszolek, Z.K.; Dickson, D.W.; Ross, O.A.; Van Deerlin, V.M.; Trojanowski, J.Q.; Hurtig, H.I.; et al. Clinical Correlations With Lewy Body Pathology inLRRK2-Related Parkinson Disease. JAMA Neurol. 2015, 72, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Poulopoulos, M.; Cortes, E.; Vonsattel, J.-P.G.; Fahn, S.; Waters, C.; Cote, L.J.; Moskowitz, C.; Honig, L.S.; Clark, L.N.; Marder, K.S.; et al. Clinical and Pathological Characteristics of LRRK2 G2019S Patients with PD. J. Mol. Neurosci. 2012, 47, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Teil, M.; Arotcarena, M.-L.; Faggiani, E.; Laferriere, F.; Bezard, E.; Dehay, B. Targeting alpha-Synuclein for PD Therapeutics: A Pursuit on All Fronts. Biomolecules 2020, 10, 391. [Google Scholar] [CrossRef]
- Elkouzi, A.; Vedam-Mai, V.; Eisinger, R.S.; Okun, M.S. Emerging therapies in Parkinson disease—Repurposed drugs and new approaches. Nat. Rev. Neurol. 2019, 15, 204–223. [Google Scholar] [CrossRef]
- Junn, E.; Lee, K.-W.; Jeong, B.S.; Chan, T.W.; Im, J.-Y.; Mouradian, M.M. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057. [Google Scholar] [CrossRef]
- Sapru, M.K.; Yates, J.W.; Hogan, S.; Jiang, L.; Halter, J.; Bohn, M.C. Silencing of human alpha-synuclein in vitro and in rat brain using lentiviral-mediated RNAi. Exp. Neurol. 2006, 198, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Doxakis, E. Post-transcriptional Regulation of alpha-Synuclein Expression by mir-7 and mir-153. J. Biol. Chem. 2010, 285, 12726–12734. [Google Scholar] [CrossRef]
- Zharikov, A.D.; Cannon, J.; Tapias, V.; Bai, Q.; Horowitz, M.P.; Shah, V.; El Ayadi, A.; Hastings, T.G.; Greenamyre, J.T.; Burton, E. shRNA targeting alpha-synuclein prevents neurodegeneration in a Parkinson’s disease model. J. Clin. Investig. 2015, 125, 2721–2735. [Google Scholar] [CrossRef]
- Recasens, A.; Perier, C.; Sue, C.M. Role of microRNAs in the Regulation of alpha-Synuclein Expression: A Systematic Review. Front. Mol. Neurosci. 2016, 9, 128. [Google Scholar] [CrossRef]
- Lewis, J.; Melrose, H.; Bumcrot, D.; Hope, A.; Zehr, C.; Lincoln, S.; Braithwaite, A.; Heather, M.; Ogholikhan, S.; Hinkle, K.; et al. In vivo silencing of alpha-synuclein using naked siRNA. Mol. Neurodegener. 2008, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Miller, A.; Lins, L.C.R.F.; Han, S.-W.; Keiser, M.S.; Boudreau, R.L.; Davidson, B.; Narayanan, N.S. RNA Interference of Human alpha-Synuclein in Mouse. Front. Neurol. 2017, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Tabrizi, S.J. Gene suppression approaches to neurodegeneration. Alzheimer’s Res. Ther. 2017, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Brys, M.; Fanning, L.; Hung, S.; Ellenbogen, A.; Penner, N.; Yang, M.; Welch, M.; Koenig, E.; David, E.; Fox, T.; et al. Randomized phase I clinical trial of anti-alpha-synuclein antibody BIIB054. Mov. Disord. 2019, 34, 1154–1163. [Google Scholar] [CrossRef]
- Jankovic, J.; Goodman, I.; Safirstein, B.; Marmon, T.K.; Schenk, D.B.; Koller, M.; Zago, W.; Ness, D.K.; Griffith, S.G.; Grundman, M.; et al. Safety and Tolerability of Multiple Ascending Doses of PRX002/RG7935, an Anti–alpha-Synuclein Monoclonal Antibody, in Patients With Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2018, 75, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Yavich, L.; Tanila, H.; Vepsäläinen, S.; Jäkälä, P. Role of alpha-Synuclein in Presynaptic Dopamine Recruitment. J. Neurosci. 2004, 24, 11165–11170. [Google Scholar] [CrossRef]
- Somayaji, M.; Cataldi, S.; Choi, S.J.; Edwards, R.H.; Mosharov, E.V.; Sulzer, D. A dual role for alpha-synuclein in facilitation and depression of dopamine release from substantia nigra neurons in vivo. Proc. Natl. Acad. Sci. USA 2020, 117, 32701–32710. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, A.; Schmitz, Y.; Fariñas, I.; Choi-Lundberg, D.; Ho, W.-H.; Castillo, P.; Shinsky, N.; García-Verdugo, J.M.; Armanini, M.; Ryan, A.; et al. Mice Lacking alpha-Synuclein Display Functional Deficits in the Nigrostriatal Dopamine System. Neuron 2000, 25, 239–252. [Google Scholar] [CrossRef]
- Kokhan, V.S.; Afanasyeva, M.A.; Van’Kin, G.I. alpha-Synuclein knockout mice have cognitive impairments. Behav. Brain Res. 2012, 231, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Benskey, M.J.; Sellnow, R.C.; Sandoval, I.M.; Sortwell, C.E.; Lipton, J.W.; Manfredsson, F.P. Silencing Alpha Synuclein in Mature Nigral Neurons Results in Rapid Neuroinflammation and Subsequent Toxicity. Front. Mol. Neurosci. 2018, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Gorbatyuk, O.S.; Li, S.; Nash, K.; Gorbatyuk, M.; Lewin, A.; Sullivan, L.F.; Mandel, R.J.; Chen, W.; Meyers, C.; Manfredsson, F.P.; et al. In Vivo RNAi-Mediated alpha-Synuclein Silencing Induces Nigrostriatal Degeneration. Mol. Ther. 2010, 18, 1450–1457. [Google Scholar] [CrossRef]
- Khodr, C.E.; Sapru, M.K.; Pedapati, J.; Han, Y.; West, N.C.; Kells, A.P.; Bankiewicz, K.S.; Bohn, M.C. An alpha-synuclein AAV gene silencing vector ameliorates a behavioral deficit in a rat model of Parkinson’s disease, but displays toxicity in dopamine neurons. Brain Res. 2011, 1395, 94–107. [Google Scholar] [CrossRef]
- Imaizumi, T.; Yamashita, K.; Taima, K.; Ishikawa, A.; Yoshida, H.; Satoh, K. Effect of peroxisome proliferator-activated receptor-gamma ligands on the expression of retinoic acid-inducible gene-I in endothelial cells stimulated with lipopolysaccharide. Prostaglandins Other Lipid Mediat. 2005, 78, 46–54. [Google Scholar] [CrossRef]
- Larsen, K.E.; Schmitz, Y.; Troyer, M.D.; Mosharov, E.; Dietrich, P.; Quazi, A.Z.; Savalle, M.; Nemani, V.; Chaudhry, F.A.; Edwards, R.H.; et al. Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J. Neurosci. 2006, 26, 11915–11922. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef]
- Nemani, V.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased Expression of alpha-Synuclein Reduces Neurotransmitter Release by Inhibiting Synaptic Vesicle Reclustering after Endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The function of alpha-synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Xu, Z.; Wu, Y.; Zhou, Y. Determining nuclear localization of alpha-synuclein in mouse brains. Neuroscience 2011, 199, 318–332. [Google Scholar] [CrossRef]
- Vivacqua, G.; Casini, A.; Vaccaro, R.; Fornai, F.; Yu, S.; D’este, L. Different sub-cellular localization of alpha-synuclein in the C57BL\6J mouse’s central nervous system by two novel monoclonal antibodies. J. Chem. Neuroanat. 2011, 41, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, V.; Mitra, J.; Wang, H.; Hegde, P.M.; Rao, K.; Hegde, M.L. A multi-faceted genotoxic network of alpha-synuclein in the nucleus and mitochondria of dopaminergic neurons in Parkinson’s disease: Emerging concepts and challenges. Prog. Neurobiol. 2020, 185, 101729. [Google Scholar] [CrossRef]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial Import and Accumulation of alpha-Synuclein Impair Complex I in Human Dopaminergic Neuronal Cultures and Parkinson Disease Brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef]
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 2010, 486, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Colla, E.; Jensen, P.H.; Pletnikova, O.; Troncoso, J.C.; Glabe, C.; Lee, M.K. Accumulation of Toxic alpha-Synuclein Oligomer within Endoplasmic Reticulum Occurs in alpha-Synucleinopathy In Vivo. J. Neurosci. 2012, 32, 3301–3305. [Google Scholar] [CrossRef]
- Thayanidhi, N.; Helm, J.R.; Nycz, D.C.; Bentley, M.; Liang, Y.; Hay, J.C. Alpha-synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol. Biol. Cell 2010, 21, 1850–1863. [Google Scholar] [CrossRef]
- Oaks, A.W.; Marsh-Armstrong, N.; Jones, J.M.; Credle, J.J.; Sidhu, A. Synucleins Antagonize Endoplasmic Reticulum Function to Modulate Dopamine Transporter Trafficking. PLoS ONE 2013, 8, e70872. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Zunke, F.; Isacson, O.; Studer, L.; Krainc, D. alpha-Synuclein–induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl. Acad. Sci. USA 2016, 113, 1931–1936. [Google Scholar] [CrossRef]
- Colla, E.; Coune, P.; Liu, Y.; Pletnikova, O.; Troncoso, J.C.; Iwatsubo, T.; Schneider, B.; Lee, M.K. Endoplasmic Reticulum Stress Is Important for the Manifestations of alpha-Synucleinopathy In Vivo. J. Neurosci. 2012, 32, 3306–3320. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli-Daley, L.A.; Gamble, K.; Schultheiss, C.E.; Riddle, D.M.; West, A.; Lee, V.M.-Y. Formation of alpha-synuclein Lewy neurite–like aggregates in axons impedes the transport of distinct endosomes. Mol. Biol. Cell 2014, 25, 4010–4023. [Google Scholar] [CrossRef] [PubMed]
- Ostrerova, N.; Petrucelli, L.; Farrer, M.; Mehta, N.; Choi, P.; Hardy, J.; Wolozin, B. alpha-Synuclein Shares Physical and Functional Homology with 14-3-3 Proteins. J. Neurosci. 1999, 19, 5782–5791. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Kalia, S.; Chau, H.; Lozano, A.; Hyman, B.T.; McLean, P. Ubiquitinylation of alpha-Synuclein by Carboxyl Terminus Hsp70-Interacting Protein (CHIP) Is Regulated by Bcl-2-Associated Athanogene 5 (BAG5). PLoS ONE 2011, 6, e14695. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, H.; McLean, P.J.; Sharma, N.; Hyman, B.T. Interaction of alpha-synuclein and synphilin-1: Effect of Parkinson’s disease-associated mutations. J. Neurochem. 2001, 77, 929–934. [Google Scholar] [CrossRef]
- Payton, J.E.; Perrin, R.J.; Clayton, D.; George, J.M. Protein–protein interactions of alpha-synuclein in brain homogenates and transfected cells. Brain Res. Mol. Brain Res. 2001, 95, 138–145. [Google Scholar] [CrossRef]
- Martinez, J.; Moeller, I.; Erdjument-Bromage, H.; Tempst, P.; Lauring, B. Parkinson’s disease-associated alpha-synuclein is a calmodulin substrate. J. Biol. Chem. 2003, 278, 17379–17387. [Google Scholar] [CrossRef]
- Leandrou, E.; Emmanouilidou, E.; Vekrellis, K. Voltage-Gated Calcium Channels and alpha-Synuclein: Implications in Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 237. [Google Scholar] [CrossRef] [PubMed]
- Khurana, V.; Peng, J.; Chung, C.Y.; Auluck, P.K.; Fanning, S.; Tardiff, D.F.; Bartels, T.; Koeva, M.; Eichhorn, S.W.; Benyamini, H.; et al. Genome-Scale Networks Link Neurodegenerative Disease Genes to alpha-Synuclein through Specific Molecular Pathways. Cell Syst. 2017, 4, 157–170.e14. [Google Scholar] [CrossRef]
- Bernal-Conde, L.D.; Ramos-Acevedo, R.; Reyes-Hernández, M.A.; Balbuena-Olvera, A.J.; Morales-Moreno, I.D.; Argüero-Sánchez, R.; Schuele, B.; Guerra-Crespo, M. Alpha-Synuclein Physiology and Pathology: A Perspective on Cellular Structures and Organelles. Front. Neurosci. 2020, 13, 1399. [Google Scholar] [CrossRef]
- Baptista, M.J.; O’Farrell, C.; Daya, S.; Ahmad, R.; Miller, D.W.; Hardy, J.; Farrer, M.; Cookson, M.R. Co-ordinate transcriptional regulation of dopamine synthesis genes by alpha-synuclein in human neuroblastoma cell lines. J. Neurochem. 2003, 85, 957–968. [Google Scholar] [CrossRef]
- Zambon, F.; Cherubini, M.; Fernandes, H.J.; Lang, C.; Ryan, B.J.; Volpato, V.; Bengoa-Vergniory, N.; Vingill, S.; Attar, M.; Booth, H.D.; et al. Cellular alpha-synuclein pathology is associated with bioenergetic dysfunction in Parkinson’s iPSC-derived dopamine neurons. Hum. Mol. Genet. 2019, 28, 2001–2013. [Google Scholar] [CrossRef]
- Surguchev, A.A.; Surguchov, A. Synucleins and Gene Expression: Ramblers in a Crowd or Cops Regulating Traffic? Front. Mol. Neurosci. 2017, 10, 224. [Google Scholar] [CrossRef] [PubMed]
- Pavlou, M.A.S.; Pinho, R.; Paiva, I.; Outeiro, T.F. The yin and yang of alpha-synuclein-associated epigenetics in Parkinson’s disease. Brain 2017, 140, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Vasudevaraju, P.; Guerrero, E.; Hegde, M.L.; Collen, T.B.; Britton, G.B.; Rao, K.S. New evidence on alpha-synuclein and Tau binding to conformation and sequence specific GC* rich DNA: Relevance to neurological disorders. J. Pharm. Bioallied. Sci. 2012, 4, 112–117. [Google Scholar]
- Padmaraju, V.; Bhaskar, J.J.; Rao, U.J.P.; Salimath, P.V.; Rao, K. Role of Advanced Glycation on Aggregation and DNA Binding Properties of alpha-Synuclein. J. Alzheimer’s Dis. 2011, 24, 211–221. [Google Scholar] [CrossRef]
- Hegde, M.L.; Rao, K.S. DNA induces folding in alpha-synuclein: Understanding the mechanism using chaperone property of osmolytes. Arch. Biochem. Biophys. 2007, 464, 57–69. [Google Scholar] [CrossRef]
- Pinho, R.; Paiva, I.; Jerčić, K.G.; Fonseca-Ornelas, L.; Gerhardt, E.; Fahlbusch, C.; Esparcia, P.G.; Kerimoglu, C.; Pavlou, M.A.S.; Villar-Piqué, A.; et al. Nuclear localization and phosphorylation modulate pathological effects of alpha-synuclein. Hum. Mol. Genet. 2019, 28, 31–50. [Google Scholar] [CrossRef]
- Schaser, A.; Osterberg, V.R.; Dent, S.E.; Stackhouse, T.L.; Wakeham, C.M.; Boutros, S.W.; Weston, L.J.; Owen, N.; Weissman, T.A.; Luna, E.; et al. Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci. Rep. 2019, 9, 1–19. [Google Scholar] [CrossRef]
- Zanzoni, A.; Marchese, D.; Agostini, F.; Bolognesi, B.; Cirillo, D.; Botta-Orfila, M.; Livi, C.M.; Rodriguez-Mulero, S.; Tartaglia, G.G. Principles of self-organization in biological pathways: A hypothesis on the autogenous association of alpha-synuclein. Nucleic Acids Res. 2013, 41, 9987–9998. [Google Scholar] [CrossRef] [PubMed]
- Berson, A.; Nativio, R.; Berger, S.L.; Bonini, N.M. Epigenetic Regulation in Neurodegenerative Diseases. Trends Neurosci. 2018, 41, 587–598. [Google Scholar] [CrossRef]
- Xylaki, M.; Atzler, B.; Outeiro, T.F. Epigenetics of the Synapse in Neurodegeneration. Curr. Neurol. Neurosci. Rep. 2019, 19, 72. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Robertson, K.D. DNA Methyltransferases, DNA Damage Repair, and Cancer. Adv. Exp. Me. Biol. 2013, 754, 3–29. [Google Scholar] [CrossRef]
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockenstein, E.; Masliah, E. Alpha-synuclein sequesters Dnmt1 from the nucleus: A novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem. 2011, 286, 9031–9037. [Google Scholar] [CrossRef]
- Goers, J.; Manning-Bog, A.B.; McCormack, A.L.; Millett, I.S.; Doniach, S.; Di Monte, D.A.; Uversky, V.N.; Fink, A.L. Nuclear Localization of alpha-Synuclein and Its Interaction with Histones†. Biochemistry 2003, 42, 8465–8471. [Google Scholar] [CrossRef]
- Jiang, P.; Gan, M.; Yen, S.-H.; McLean, P.; Dickson, D.W. Histones facilitate alpha-synuclein aggregation during neuronal apoptosis. Acta Neuropathol. 2017, 133, 547–558. [Google Scholar] [CrossRef]
- Mazzocchi, M.; Collins, L.M.; Sullivan, A.M.; O’Keeffe, G.W. The class II histone deacetylases as therapeutic targets for Parkinson’s disease. Neuronal Signal. 2020, 4, NS20200001. [Google Scholar] [CrossRef] [PubMed]
- Harrison, I.F.; Smith, A.D.; Dexter, D.T. Pathological histone acetylation in Parkinson’s disease: Neuroprotection and inhibition of microglial activation through SIRT 2 inhibition. Neurosci. Lett. 2018, 666, 48–57. [Google Scholar] [CrossRef]
- Park, G.; Tan, J.; Garcia, G.; Kang, Y.; Salvesen, G.; Zhang, Z. Regulation of Histone Acetylation by Autophagy in Parkinson Disease. J. Biol. Chem. 2016, 291, 3531–3540. [Google Scholar] [CrossRef]
- Gebremedhin, K.G.; Rademacher, D.J. Histone H3 acetylation in the postmortem Parkinson’s disease primary motor cortex. Neurosci. Lett. 2016, 627, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Kontopoulos, E.; Parvin, J.D.; Feany, M.B. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 2006, 15, 3012–3023. [Google Scholar] [CrossRef] [PubMed]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.C.; McLean, P.J.; et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef]
- Paiva, I.; Pinho, R.; Pavlou, M.A.; Hennion, M.; Wales, P.; Schütz, A.-L.; Rajput, A.; Szegő, É.M.; Kerimoglu, C.; Gerhardt, E.; et al. Sodium butyrate rescues dopaminergic cells from alpha-synuclein-induced transcriptional deregulation and DNA damage. Hum. Mol. Genet. 2017, 26, 2231–2246. [Google Scholar] [CrossRef] [PubMed]
- Mazzocchi, M.; Wyatt, S.L.; Mercatelli, D.; Morari, M.; Morales-Prieto, N.; Collins, L.; Sullivan, A.M.; O’Keeffe, G.W. Gene Co-expression Analysis Identifies Histone Deacetylase 5 and 9 Expression in Midbrain Dopamine Neurons and as Regulators of Neurite Growth via Bone Morphogenetic Protein Signaling. Front. Cell Dev. Biol. 2019, 7, 191. [Google Scholar] [CrossRef] [PubMed]
- Harrison, I.F.; Powell, N.M.; Dexter, D.T. The histone deacetylase inhibitor nicotinamide exacerbates neurodegeneration in the lactacystin rat model of Parkinson’s disease. J. Neurochem. 2019, 148, 136–156. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Y.; Zhu, K.; Chi, S.; Wang, C.; Xie, A. Emerging Role of Sirtuin 2 in Parkinson’s Disease. Front. Aging Neurosci. 2020, 11, 372. [Google Scholar] [CrossRef]
- Zhang, Y.; Kwon, S.; Yamaguchi, T.; Cubizolles, F.; Rousseaux, S.; Kneissel, M.; Cao, C.; Li, N.; Cheng, H.-L.; Chua, K.; et al. Mice Lacking Histone Deacetylase 6 Have Hyperacetylated Tubulin but Are Viable and Develop Normally. Mol. Cell. Biol. 2008, 28, 1688–1701. [Google Scholar] [CrossRef]
- Cook, C.; Stankowski, J.N.; Carlomagno, Y.; Stetler, C.; Petrucelli, L. Acetylation: A new key to unlock tau’s role in neurodegeneration. Alzheimer’s Res. Ther. 2014, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Kanthasamy, A.; Ghosh, A.; Yang, Y.; Anantharam, V.; Kanthasamy, A.G. alpha-Synuclein negatively regulates protein kinase Cdelta expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. J. Neurosci. 2011, 31, 2035–2051. [Google Scholar] [CrossRef]
- Chen, K.; Bennett, S.A.; Rana, N.; Yousuf, H.; Said, M.; Taaseen, S.; Mendo, N.; Meltser, S.M.; Torrente, M.P. Neurodegenerative Disease Proteinopathies Are Connected to Distinct Histone Post-translational Modification Landscapes. ACS Chem. Neurosci. 2018, 9, 838–848. [Google Scholar] [CrossRef]
- Sugeno, N.; Jäckel, S.; Voigt, A.; Wassouf, Z.; Schulze-Hentrich, J.; Kahle, P.J. alpha-Synuclein enhances histone H3 lysine-9 dimethylation and H3K9me2-dependent transcriptional responses. Sci. Rep. 2016, 6, 36328. [Google Scholar] [CrossRef]
- Davidi, D.; Schechter, M.; Elhadi, S.A.; Matatov, A.; Nathanson, L.; Sharon, R. α-Synuclein Translocates to the Nucleus to Activate Retinoic-Acid-Dependent Gene Transcription. iScience 2020, 23, 100910. [Google Scholar] [CrossRef]
- Yakunin, E.; Loeb, V.; Kisos, H.; Biala, Y.; Yehuda, S.; Yaari, Y.; Selkoe, D.J.; Sharon, R. Alpha-synuclein neuropathology is controlled by nuclear hormone receptors and enhanced by docosahexaenoic acid in a mouse model for Parkinson’s disease. Brain Pathol. 2012, 22, 280–294. [Google Scholar] [CrossRef]
- Yakunin, E.; Kisos, H.; Kulik, W.; Grigoletto, J.; Wanders, R.J.; Sharon, R. The regulation of catalase activity by PPAR gamma is affected by alpha-synuclein. Ann. Clin. Transl. Neurol. 2014, 1, 145–159. [Google Scholar] [CrossRef]
- Volakakis, N.; Tiklova, K.; Decressac, M.; Papathanou, M.; Mattsson, B.; Gillberg, L.; Nobre, A.; Björklund, A.; Perlmann, T. Nurr1 and Retinoid X Receptor Ligands Stimulate Ret Signaling in Dopamine Neurons and Can Alleviate alpha-Synuclein Disrupted Gene Expression. J. Neurosci. 2015, 35, 14370–14385. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Le, W.; Kompoliti, K.; Jankovic, J.; Mufson, E.J.; Kordower, J.H. Nurr1 in Parkinson’s disease and related disorders. J. Comp. Neurol. 2006, 494, 495–514. [Google Scholar] [CrossRef]
- Decressac, M.; Volakakis, N.; Björklund, A.; Perlmann, T. NURR1 in Parkinson disease—from pathogenesis to therapeutic potential. Nat. Rev. Neurol. 2013, 9, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Spathis, A.D.; Asvos, X.; Ziavra, D.; Karampelas, T.; Topouzis, S.; Cournia, Z.; Qing, X.; Alexakos, P.; Smits, L.M.; Dalla, C.; et al. Nurr1:RXRalpha heterodimer activation as monotherapy for Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 3999–4004. [Google Scholar] [CrossRef]
- Siddiqui, A.; Chinta, S.J.; Mallajosyula, J.K.; Rajagopolan, S.; Hanson, I.; Rane, A.; Melov, S.; Andersen, J.K. Selective binding of nuclear alpha-synuclein to the PGC1alpha promoter under conditions of oxidative stress may contribute to losses in mitochondrial function: Implications for Parkinson’s disease. Free. Radic. Biol. Med. 2012, 53, 993–1003. [Google Scholar] [CrossRef]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef]
- Eschbach, J.; von Einem, B.; Müller, K.; Bayer, H.; Scheffold, A.; Morrison, B.E.; Rudolph, K.L.; Thal, D.R.; Witting, A.; Weydt, P.; et al. Mutual exacerbation of peroxisome proliferator-activated receptor gamma coactivator 1alpha deregulation and alpha-synuclein oligomerization. Ann. Neurol. 2015, 77, 15–32. [Google Scholar] [CrossRef]
- Kim, S.Y.; Choi, K.C.; Chang, M.S.; Kim, M.H.; Na, Y.-S.; Lee, J.E.; Jin, B.K.; Lee, B.-H.; Baik, J.-H. The Dopamine D2 Receptor Regulates the Development of Dopaminergic Neurons via Extracellular Signal-Regulated Kinase and Nurr1 Activation. J. Neurosci. 2006, 26, 4567–4576. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Burgess, J.D.; Faroqi, A.; DeMeo, N.N.; Fiesel, F.C.; Springer, W.; Delenclos, M.; McLean, P.J. Alpha-synuclein-induced mitochondrial dysfunction is mediated via a sirtuin 3-dependent pathway. Mol. Neurodegener. 2020, 15, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Bause, A.S.; Haigis, M.C. SIRT3 regulation of mitochondrial oxidative stress. Exp. Gerontol. 2013, 48, 634–639. [Google Scholar] [CrossRef]
- Sidorova-Darmos, E.; Sommer, R.; Eubanks, J.H. The Role of SIRT3 in the Brain Under Physiological and Pathological Conditions. Front. Cell. Neurosci. 2018, 12, 196. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a New Target of PGC-1alpha, Plays an Important Role in the Suppression of ROS and Mitochondrial Biogenesis. PLoS ONE 2010, 5, e11707. [Google Scholar] [CrossRef] [PubMed]
- Iwata, A.; Miura, S.; Kanazawa, I.; Sawada, M.; Nukina, N. alpha-Synuclein forms a complex with transcription factor Elk-1. J. Neurochem. 2001, 77, 239–252. [Google Scholar] [CrossRef]
- Hashimoto, M.; Takenouchi, T.; Rockenstein, E.; Masliah, E. Alpha-synuclein up-regulates expression of caveolin-1 and down-regulates extracellular signal-regulated kinase activity in B103 neuroblastoma cells: Role in the pathogenesis of Parkinson’s disease. J. Neurochem. 2003, 85, 1468–1479. [Google Scholar] [CrossRef] [PubMed]
- Besnard, A.; Galan-Rodriguez, B.; Vanhoutte, P.; Caboche, J. Elk-1 a Transcription Factor with Multiple Facets in the Brain. Front. Neurosci. 2011, 5, 35. [Google Scholar] [CrossRef]
- Tolö, J.; Taschenberger, G.; Leite, K.; Stahlberg, M.A.; Spehlbrink, G.; Kues, J.; Munari, F.; Capaldi, S.; Becker, S.; Zweckstetter, M.; et al. Pathophysiological Consequences of Neuronal alpha-Synuclein Overexpression: Impacts on Ion Homeostasis, Stress Signaling, Mitochondrial Integrity, and Electrical Activity. Front. Mol. Neurosci. 2018, 11, 49. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Wang, X.-Y.; Kang, W.-Y.; Zhang, Y.; Zhang, Y.; Zhou, Y.; Quinn, T.; Liu, J.; Chen, S.-D. Extracellular signal-regulated kinase is involved in alpha-synuclein-induced mitochondrial dynamic disorders by regulating dynamin-like protein 1. Neurobiol. Aging 2012, 33, 2841–2854. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.H.; Rah, J.C.; Choi, S.H.; Shin, J.K.; Min, K.; Kim, H.S.; Park, C.H.; Kim, S.; Kim, E.M.; Lee, S.H.; et al. Alpha-synuclein regulates neuronal survival via Bcl-2 family expression and PI3/Akt kinase pathway. FASEB J. 2002, 16, 1826–1828. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Cohen, S.; Li, B.; Tsien, R.W. Exploring the dominant role of Cav1 channels in signalling to the nucleus. Biosci. Rep. 2012, 33, 97–101. [Google Scholar] [CrossRef]
- Li, B.; Tadross, M.R.; Tsien, R.W. Sequential ionic and conformational signaling by calcium channels drives neuronal gene expression. Science 2016, 351, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Lenaeus, M.J.; El-Din, T.M.G. Structure and Pharmacology of Voltage-Gated Sodium and Calcium Channels. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 133–154. [Google Scholar] [CrossRef]
- Striessnig, J.; Pinggera, A.; Kaur, G.; Bock, G.; Tuluc, P. L-type Ca(2+) channels in heart and brain. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2014, 3, 15–38. [Google Scholar] [CrossRef]
- Ben-Johny, M.; Yue, D.T. Calmodulin regulation (calmodulation) of voltage-gated calcium channels. J. Gen. Physiol. 2014, 143, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Johny, M.; Yang, P.S.; Bazzazi, H.; Yue, D.T. Dynamic switching of calmodulin interactions underlies Ca2+ regulation of CaV1.3 channels. Nat. Commun. 2013, 4, 1717. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.M.; Li, B.; Tsien, R.W.; Ma, H. Evolutionary and functional perspectives on signaling from neuronal surface to nucleus. Biochem. Biophys. Res. Commun. 2015, 460, 88–99. [Google Scholar] [CrossRef][Green Version]
- Bito, H.; Deisseroth, K.; Tsien, R.W. CREB Phosphorylation and Dephosphorylation: A Ca2+- and Stimulus Duration–Dependent Switch for Hippocampal Gene Expression. Cell 1996, 87, 1203–1214. [Google Scholar] [CrossRef]
- Wu, H.; Peisley, A.; Graef, I.A.; Crabtree, G.R. NFAT signaling and the invention of vertebrates. Trends Cell Biol. 2007, 17, 251–260. [Google Scholar] [CrossRef]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef]
- Macian, F. NFAT proteins: Key regulators of T-cell development and function. Nat. Rev. Immunol. 2005, 5, 472–484. [Google Scholar] [CrossRef]
- Horsley, V.; Pavlath, G.K. NFAT: Ubiquitous regulator of cell differentiation and adaptation. J. Cell Biol. 2002, 156, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.Y.; Khurana, V.; Yi, S.; Sahni, N.; Loh, K.H.; Auluck, P.K.; Baru, V.; Udeshi, N.D.; Freyzon, Y.; Carr, S.A.; et al. In Situ Peroxidase Labeling and Mass-Spectrometry Connects Alpha-Synuclein Directly to Endocytic Trafficking and mRNA Metabolism in Neurons. Cell Syst. 2017, 4, 242–250.e4. [Google Scholar] [CrossRef]
- Luo, J.; Sun, L.; Lin, X.; Liu, G.; Yu, J.; Parisiadou, L.; Xie, C.; Ding, J.; Cai, H. A calcineurin- and NFAT-dependent pathway is involved in alpha-synuclein-induced degeneration of midbrain dopaminergic neurons. Hum. Mol. Genet. 2014, 23, 6567–6574. [Google Scholar] [CrossRef]
- Shi, X.; Sun, Y.; Wang, P.; Gu, L.; Wang, L.; Yang, H.; Wei, Q.; Li, Z.; Luo, J. The interaction between calcineurin and alpha-synuclein is regulated by calcium and calmodulin. Biochem. Biophys. Res. Commun. 2018, 496, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; De Silva, H.R.; Kittel, A.; Saitoh, T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef]
- Baltic, S.; Perovic, M.; Mladenovic, A.; Raicevic, N.; Ruzdijic, S.; Rakic, L.; Kanazir, S. Alpha-synuclein is expressed in different tissues during human fetal development. J. Mol. Neurosci. 2004, 22, 199–204. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Somayaji, M.; Lanseur, Z.; Choi, S.J.; Sulzer, D.; Mosharov, E.V. Roles for α-Synuclein in Gene Expression. Genes 2021, 12, 1166. https://doi.org/10.3390/genes12081166
Somayaji M, Lanseur Z, Choi SJ, Sulzer D, Mosharov EV. Roles for α-Synuclein in Gene Expression. Genes. 2021; 12(8):1166. https://doi.org/10.3390/genes12081166
Chicago/Turabian StyleSomayaji, Mahalakshmi, Zina Lanseur, Se Joon Choi, David Sulzer, and Eugene V. Mosharov. 2021. "Roles for α-Synuclein in Gene Expression" Genes 12, no. 8: 1166. https://doi.org/10.3390/genes12081166
APA StyleSomayaji, M., Lanseur, Z., Choi, S. J., Sulzer, D., & Mosharov, E. V. (2021). Roles for α-Synuclein in Gene Expression. Genes, 12(8), 1166. https://doi.org/10.3390/genes12081166