The Biochemistry of Cytoplasmic Incompatibility Caused by Endosymbiotic Bacteria


Abstract
1. Introduction
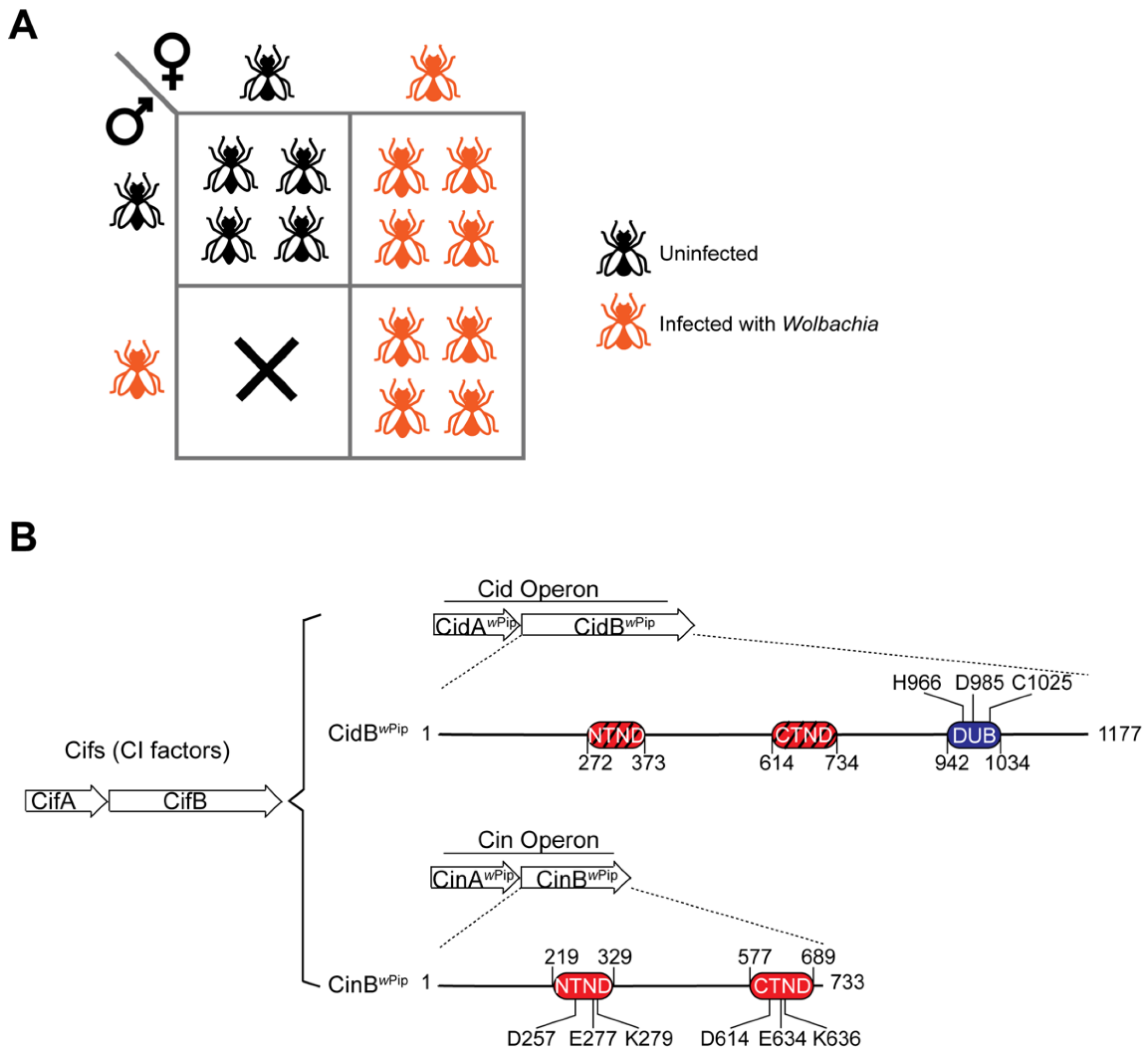
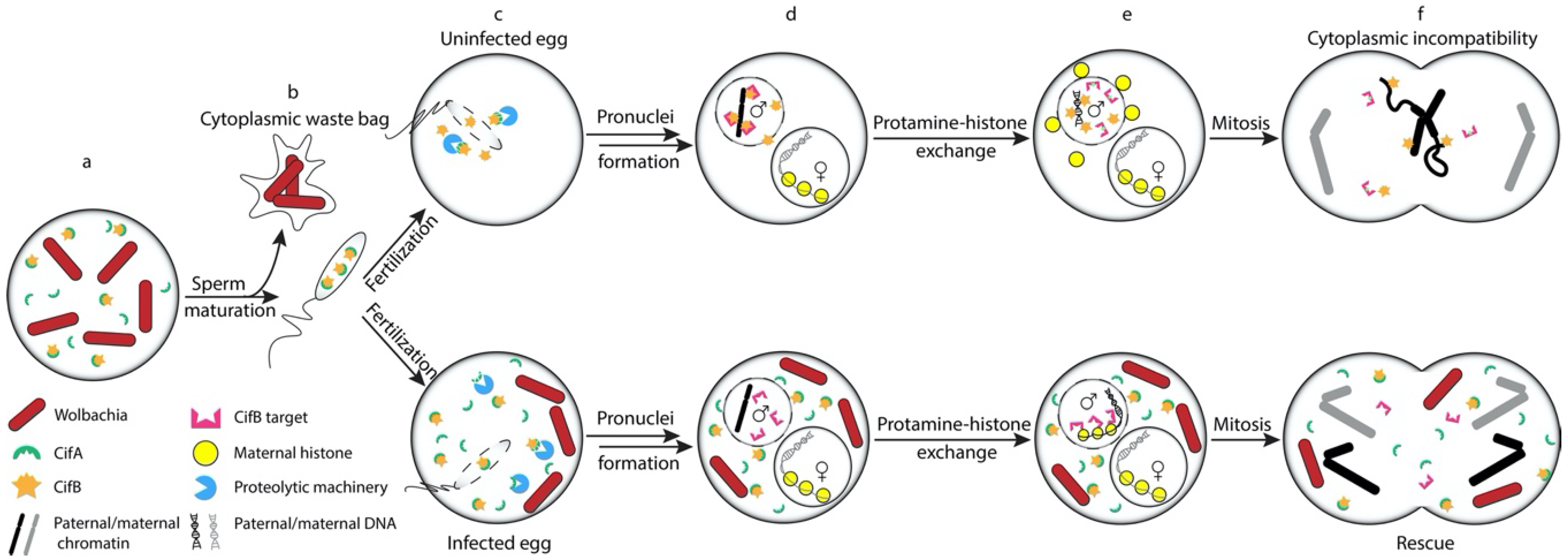
2. Cell Biology of Cytoplasmic Incompatibility
3. The Wolbachia CI Factors (Cifs)
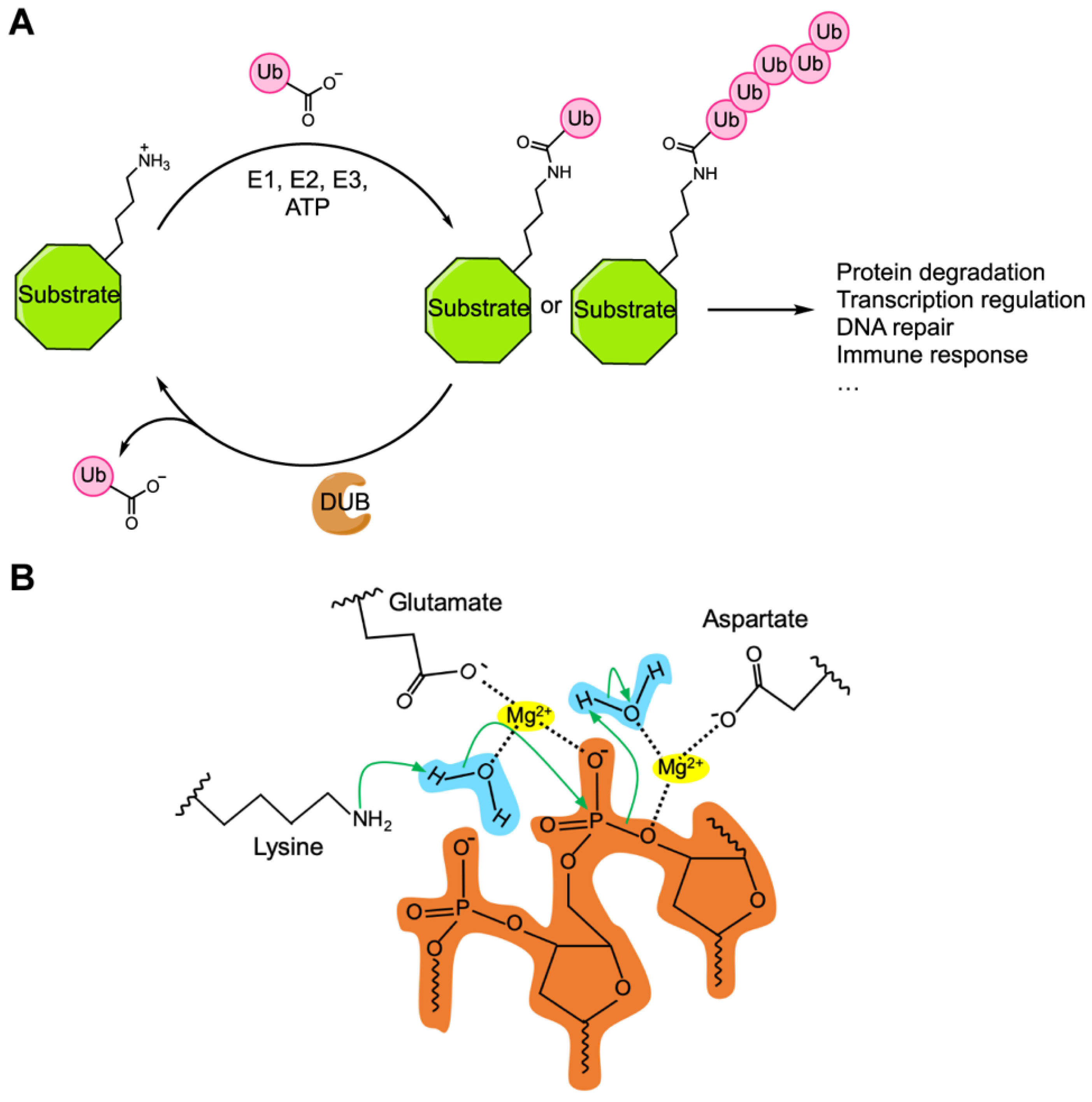
4. Sequence Motifs and Biochemical Activities of the Cif Proteins
5. CidA–CidB, a CI Inducing Protein Pair Containing Deubiquitylase Activity
6. CinA–CinB, a CI Inducing Protein Pair Containing Nuclease Activity
7. Models of CI Induction and Rescue
8. Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hurst, G.D.; Frost, C.L. Reproductive parasitism: Maternally inherited symbionts in a biparental world. Cold Spring Harb. Perspect. Biol. 2015, 7, a017699. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.J.; Hoerauf, A. Wolbachia bacteria of filarial nematodes. Parasitol. Today 1999, 15, 437–442. [Google Scholar] [CrossRef]
- Werren, J.H.; Windsor, D.M. Wolbachia infection frequencies in insects: Evidence of a global equilibrium? Proc. Biol. Sci. 2000, 267, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Hilgenboecker, K.; Hammerstein, P.; Schlattmann, P.; Telschow, A.; Werren, J.H. How many species are infected with Wolbachia?—A statistical analysis of current data. FEMS Microbiol. Lett. 2008, 281, 215–220. [Google Scholar] [CrossRef]
- Hoffmann, A.A.; Turelli, M.; Harshman, L.G. Factors affecting the distribution of cytoplasmic incompatibility in Drosophila simulans. Genetics 1990, 126, 933–948. [Google Scholar]
- Bressac, C.; Rousset, F. The reproductive incompatibility system in Drosophila simulans: DAPI-staining analysis of the Wolbachia symbionts in sperm cysts. J. Invertebr. Pathol. 1993, 61, 226–230. [Google Scholar] [CrossRef]
- Dedeine, F.; Vavre, F.; Fleury, F.; Loppin, B.; Hochberg, M.E.; Bouletreau, M. Removing symbiotic Wolbachia bacteria specifically inhibits oogenesis in a parasitic wasp. Proc. Natl. Acad. Sci. USA 2001, 98, 6247–6252. [Google Scholar] [CrossRef]
- Taylor, M.J.; Voronin, D.; Johnston, K.L.; Ford, L. Wolbachia filarial interactions. Cell Microbiol. 2013, 15, 520–526. [Google Scholar] [CrossRef]
- Serbus, L.R.; Casper-Lindley, C.; Landmann, F.; Sullivan, W. The genetics and cell biology of Wolbachia-host interactions. Annu. Rev. Genet. 2008, 42, 683–707. [Google Scholar] [CrossRef]
- Werren, J.H.; Baldo, L.; Clark, M.E. Wolbachia: Master manipulators of invertebrate biology. Nat. Rev. Microbiol. 2008, 6, 741–751. [Google Scholar] [CrossRef]
- Laven, H. Reziprok unterschiedliche Kreuzbarkeit von Stechmücken (Culicidae) und ihre Deutung als plasmatische Vererbung. Z. Indukt. Abstamm. Vererbungslehre 1953, 85, 118–136. [Google Scholar] [CrossRef]
- Yen, J.H.; Barr, A.R. The etiological agent of cytoplasmic incompatibility in Culex pipiens. J. Invertebr. Pathol. 1973, 22, 242–250. [Google Scholar] [CrossRef]
- Lassy, C.W.; Karr, T.L. Cytological analysis of fertilization and early embryonic development in incompatible crosses of Drosophila simulans. Mech. Dev. 1996, 57, 47–58. [Google Scholar] [CrossRef]
- Tram, U.; Sullivan, W. Role of delayed nuclear envelope breakdown and mitosis in Wolbachia-induced cytoplasmic incompatibility. Science 2002, 296, 1124–1126. [Google Scholar] [CrossRef] [PubMed]
- Bordenstein, S.R.; Uy, J.J.; Werren, J.H. Host Genotype Determines Cytoplasmic Incompatibility Type in the Haplodiploid Genus Nasonia. Genetics 2003, 164, 223–233. [Google Scholar]
- Xi, Z.; Khoo, C.C.; Dobson, S.L. Wolbachia establishment and invasion in an Aedes aegypti laboratory population. Science 2005, 310, 326–328. [Google Scholar] [CrossRef]
- Zabalou, S.; Riegler, M.; Theodorakopoulou, M.; Stauffer, C.; Savakis, C.; Bourtzis, K. Wolbachia-induced cytoplasmic incompatibility as a means for insect pest population control. Proc. Natl. Acad. Sci. USA 2004, 101, 15042–15045. [Google Scholar] [CrossRef]
- Flores, H.A.; O’Neill, S.L. Controlling vector-borne diseases by releasing modified mosquitoes. Nat. Rev. Microbiol. 2018, 16, 508–518. [Google Scholar] [CrossRef]
- Moreira, L.A.; Iturbe-Ormaetxe, I.; Jeffery, J.A.; Lu, G.; Pyke, A.T.; Hedges, L.M.; Rocha, B.C.; Hall-Mendelin, S.; Day, A.; Riegler, M.; et al. A Wolbachia symbiont in Aedes aegypti limits infection with dengue, Chikungunya, and Plasmodium. Cell 2009, 139, 1268–1278. [Google Scholar] [CrossRef]
- Hoffmann, A.A.; Montgomery, B.L.; Popovici, J.; Iturbe-Ormaetxe, I.; Johnson, P.H.; Muzzi, F.; Greenfield, M.; Durkan, M.; Leong, Y.S.; Dong, Y.; et al. Successful establishment of Wolbachia in Aedes populations to suppress dengue transmission. Nature 2011, 476, 454–457. [Google Scholar] [CrossRef]
- Callaway, E. Rio fights Zika with biggest release yet of bacteria-infected mosquitoes. Nature 2016, 539, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhang, D.; Li, Y.; Yang, C.; Wu, Y.; Liang, X.; Liang, Y.; Pan, X.; Hu, L.; Sun, Q.; et al. Incompatible and sterile insect techniques combined eliminate mosquitoes. Nature 2019, 572, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Werren, J.H. Biology of Wolbachia. Annu. Rev. Entomol. 1997, 42, 587–609. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, J.F.; Ronau, J.A.; Hochstrasser, M. A Wolbachia deubiquitylating enzyme induces cytoplasmic incompatibility. Nat. Microbiol. 2017, 2, 17007. [Google Scholar] [CrossRef] [PubMed]
- LePage, D.P.; Metcalf, J.A.; Bordenstein, S.R.; On, J.; Perlmutter, J.I.; Shropshire, J.D.; Layton, E.M.; Funkhouser-Jones, L.J.; Beckmann, J.F.; Bordenstein, S.R. Prophage WO genes recapitulate and enhance Wolbachia-induced cytoplasmic incompatibility. Nature 2017, 543, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, T.; Noda, H.; Ito, S. Cardinium symbionts cause cytoplasmic incompatibility in spider mites. Heredity 2007, 98, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Loppin, B.; Bonnefoy, E.; Anselme, C.; Laurencon, A.; Karr, T.L.; Couble, P. The histone H3.3 chaperone HIRA is essential for chromatin assembly in the male pronucleus. Nature 2005, 437, 1386–1390. [Google Scholar] [CrossRef]
- Balhorn, R. The protamine family of sperm nuclear proteins. Genome Biol. 2007, 8, 1–8. [Google Scholar] [CrossRef]
- Landmann, F.; Orsi, G.A.; Loppin, B.; Sullivan, W. Wolbachia-mediated cytoplasmic incompatibility is associated with impaired histone deposition in the male pronucleus. PLoS Pathog. 2009, 5, e1000343. [Google Scholar] [CrossRef]
- Loppin, B.; Dubruille, R.; Horard, B. The intimate genetics of Drosophila fertilization. Open Biol. 2015, 5, 150076. [Google Scholar] [CrossRef]
- Reed, K.M.; Werren, J.H. Induction of paternal genome loss by the paternal-sex-ratio chromosome and cytoplasmic incompatibility bacteria (Wolbachia): A comparative study of early embryonic events. Mol. Reprod. Dev. 1995, 40, 408–418. [Google Scholar] [CrossRef]
- Callaini, G.; Dallai, R.; Riparbelli, M.G. Wolbachia-induced delay of paternal chromatin condensation does not prevent maternal chromosomes from entering anaphase in incompatible crosses of Drosophila simulans. J. Cell Sci. 1997, 110, 271–280. [Google Scholar]
- Tram, U.; Fredrick, K.; Werren, J.H.; Sullivan, W. Paternal chromosome segregation during the first mitotic division determines Wolbachia-induced cytoplasmic incompatibility phenotype. J. Cell Sci. 2006, 119, 3655–3663. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, J.F.; Fallon, A.M. Detection of the Wolbachia protein WPIP0282 in mosquito spermathecae: Implications for cytoplasmic incompatibility. Insect Biochem. Mol. Biol. 2013, 43, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Shropshire, J.D.; On, J.; Layton, E.M.; Zhou, H.; Bordenstein, S.R. One prophage WO gene rescues cytoplasmic incompatibility in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2018, 115, 4987–4991. [Google Scholar] [CrossRef] [PubMed]
- Bing, X.L.; Zhao, D.S.; Sun, J.T.; Zhang, K.J.; Hong, X.Y. Genomic Analysis of Wolbachia from Laodelphax striatellus (Delphacidae, Hemiptera) Reveals Insights into Its “Jekyll and Hyde” Mode of Infection Pattern. Genome Biol. Evol. 2020, 12, 3818–3831. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, A.R.I.; Rice, D.W.; Bordenstein, S.R.; Brooks, A.W.; Bordenstein, S.R.; Newton, I.L.G. Evolutionary Genetics of Cytoplasmic Incompatibility Genes cifA and cifB in Prophage WO of Wolbachia. Genome Biol. Evol. 2018, 10, 434–451. [Google Scholar] [CrossRef]
- Beckmann, J.F.; Bonneau, M.; Chen, H.; Hochstrasser, M.; Poinsot, D.; Mercot, H.; Weill, M.; Sicard, M.; Charlat, S. The Toxin-Antidote Model of Cytoplasmic Incompatibility: Genetics and Evolutionary Implications. Trends Genet. 2019, 35, 175–185. [Google Scholar] [CrossRef]
- Chen, H.; Ronau, J.A.; Beckmann, J.F.; Hochstrasser, M. A Wolbachia nuclease and its binding partner provide a distinct mechanism for cytoplasmic incompatibility. Proc. Natl. Acad. Sci. USA 2019, 116, 22314–22321. [Google Scholar] [CrossRef]
- Beckmann, J.F.; Sharma, G.D.; Mendez, L.; Chen, H.; Hochstrasser, M. The Wolbachia cytoplasmic incompatibility enzyme CidB targets nuclear import and protamine-histone exchange factors. Elife 2019, 8, e50026. [Google Scholar] [CrossRef]
- Shropshire, J.D.; Bordenstein, S.R. Two-By-One model of cytoplasmic incompatibility: Synthetic recapitulation by transgenic expression of cifA and cifB in Drosophila. PLoS Genet. 2019, 15, e1008221. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, M.; Atyame, C.; Beji, M.; Justy, F.; Cohen-Gonsaud, M.; Sicard, M.; Weill, M. Culex pipiens crossing type diversity is governed by an amplified and polymorphic operon of Wolbachia. Nat. Commun. 2018, 9, 319. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, M.; Caputo, B.; Ligier, A.; Caparros, R.; Unal, S.; Perriat-Sanguinet, M.; Arnoldi, D.; Sicard, M.; Weill, M. Variation in Wolbachia cidB gene, but not cidA, is associated with cytoplasmic incompatibility mod phenotype diversity in Culex pipiens. Mol. Ecol. 2019, 28, 4725–4736. [Google Scholar] [CrossRef] [PubMed]
- Cooper, B.S.; Vanderpool, D.; Conner, W.R.; Matute, D.R.; Turelli, M. Wolbachia Acquisition by Drosophila yakuba-clade Hosts and Transfer of Incompatibility Loci Between Distantly Related Wolbachia. Genetics 2019, 212, 1399–1419. [Google Scholar] [CrossRef] [PubMed]
- Meany, M.K.; Conner, W.R.; Richter, S.V.; Bailey, J.A.; Turelli, M.; Cooper, B.S. Loss of cytoplasmic incompatibility and minimal fecundity effects explain relatively low Wolbachia frequencies in Drosophila mauritiana. Evolution 2019, 73, 1278–1295. [Google Scholar] [CrossRef]
- Bordenstein, S.R.; Bordenstein, S.R. Eukaryotic association module in phage WO genomes from Wolbachia. Nat. Commun. 2016, 7, 13155. [Google Scholar] [CrossRef]
- Masui, S.; Kuroiwa, H.; Sasaki, T.; Inui, M.; Kuroiwa, T.; Ishikawa, H. Bacteriophage WO and virus-like particles in Wolbachia, an endosymbiont of arthropods. Biochem. Biophys. Res. Commun. 2001, 283, 1099–1104. [Google Scholar] [CrossRef]
- Morrow, J.L.; Schneider, D.I.; Klasson, L.; Janitz, C.; Miller, W.J.; Riegler, M. Parallel Sequencing of Wolbachia wCer2 from Donor and Novel Hosts Reveals Multiple Incompatibility Factors and Genome Stability after Host Transfers. Genome Biol. Evol. 2020, 12, 720–735. [Google Scholar] [CrossRef]
- Gillespie, J.J.; Driscoll, T.P.; Verhoeve, V.I.; Rahman, M.S.; Macaluso, K.R.; Azad, A.F. A Tangled Web: Origins of Reproductive Parasitism. Genome Biol. Evol. 2018, 10, 2292–2309. [Google Scholar] [CrossRef]
- Shropshire, J.D.; Kalra, M.; Bordenstein, S.R. Evolution-guided mutagenesis of the cytoplasmic incompatibility proteins: Identifying 1 CifA’s complex functional repertoire and new essential regions in CifB. BioRxiv 2020. [Google Scholar] [CrossRef]
- Reverter, D.; Lima, C.D. A basis for SUMO protease specificity provided by analysis of human Senp2 and a Senp2-SUMO complex. Structure 2004, 12, 1519–1531. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef]
- Haglund, K.; Dikic, I. Ubiquitylation and cell signaling. EMBO J. 2005, 24, 3353–3359. [Google Scholar] [CrossRef]
- Park, Y.; Jin, H.S.; Aki, D.; Lee, J.; Liu, Y.C. The ubiquitin system in immune regulation. Adv. Immunol. 2014, 124, 17–66. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The Ubiquitin-Proteasome Proteolytic Pathway: Destruction for the Sake of Construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Lin, Y.H.; Machner, M.P. Exploitation of the host cell ubiquitin machinery by microbial effector proteins. J. Cell Sci. 2017, 130, 1985–1996. [Google Scholar] [CrossRef]
- Hermanns, T.; Hofmann, K. Bacterial DUBs: Deubiquitination beyond the seven classes. Biochem. Soc. Trans. 2019, 47, 1857–1866. [Google Scholar] [CrossRef]
- Pruneda, J.N.; Durkin, C.H.; Geurink, P.P.; Ovaa, H.; Santhanam, B.; Holden, D.W.; Komander, D. The Molecular Basis for Ubiquitin and Ubiquitin-like Specificities in Bacterial Effector Proteases. Mol. Cell 2016, 63, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Le Negrate, G.; Krieg, A.; Faustin, B.; Loeffler, M.; Godzik, A.; Krajewski, S.; Reed, J.C. ChlaDub1 of Chlamydia trachomatis suppresses NF-κB activation and inhibits IκBα ubiquitination and degradation. Cell Microbiol. 2008, 10, 1879–1892. [Google Scholar] [CrossRef] [PubMed]
- Sheedlo, M.J.; Qiu, J.; Tan, Y.; Paul, L.N.; Luo, Z.Q.; Das, C. Structural basis of substrate recognition by a bacterial deubiquitinase important for dynamics of phagosome ubiquitination. Proc. Natl. Acad. Sci. USA 2015, 112, 15090–15095. [Google Scholar] [CrossRef] [PubMed]
- Ronau, J.A.; Beckmann, J.F.; Hochstrasser, M. Substrate specificity of the ubiquitin and Ubl proteases. Cell Res. 2016, 26, 441–456. [Google Scholar] [CrossRef] [PubMed]
- Kunz, K.; Piller, T.; Muller, S. SUMO-specific proteases and isopeptidases of the SENP family at a glance. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Pruneda, J.N.; Bastidas, R.J.; Bertsoulaki, E.; Swatek, K.N.; Santhanam, B.; Clague, M.J.; Valdivia, R.H.; Urbe, S.; Komander, D. A Chlamydia effector combining deubiquitination and acetylation activities induces Golgi fragmentation. Nat. Microbiol. 2018, 3, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Keitany, G.; Li, Y.; Wang, Y.; Ball, H.L.; Goldsmith, E.J.; Orth, K. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 2006, 312, 1211–1214. [Google Scholar] [CrossRef]
- Berk, J.M.; Lim, C.; Ronau, J.A.; Chaudhuri, A.; Chen, H.; Beckmann, J.F.; Loria, J.P.; Xiong, Y.; Hochstrasser, M. A deubiquitylase with an unusually high-affinity ubiquitin-binding domain from the scrub typhus pathogen Orientia tsutsugamushi. Nat. Commun. 2020, 11, 2343. [Google Scholar] [CrossRef]
- Presgraves, D.C. A genetic test of the mechanism of Wolbachia-induced cytoplasmic incompatibility in Drosophila. Genetics 2000, 154, 771–776. [Google Scholar]
- Liu, P.; Gan, W.; Su, S.; Hauenstein, A.V.; Fu, T.-M.; Brasher, B.; Schwerdtfeger, C.; Liang, A.C.; Xu, M.; Wei, W. K63-linked polyubiquitin chains bind to DNA to facilitate DNA damage repair. Sci. Signal. 2018, 11, eaar8133. [Google Scholar] [CrossRef]
- Schwertman, P.; Bekker-Jensen, S.; Mailand, N. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat. Rev. Mol. Cell Biol. 2016, 17, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Emelyanov, A.V.; Rabbani, J.; Mehta, M.; Vershilova, E.; Keogh, M.C.; Fyodorov, D.V. Drosophila TAP/p32 is a core histone chaperone that cooperates with NAP-1, NLP, and nucleophosmin in sperm chromatin remodeling during fertilization. Genes Dev. 2014, 28, 2027–2040. [Google Scholar] [CrossRef] [PubMed]
- Bing, X.L.; Lu, Y.J.; Xia, C.B.; Xia, X.; Hong, X.Y. Transcriptome of Tetranychus urticae embryos reveals insights into Wolbachia-induced cytoplasmic incompatibility. Insect Mol. Biol. 2020, 29, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Knizewski, L.; Kinch, L.N.; Grishin, N.V.; Rychlewski, L.; Ginalski, K. Realm of PD-(D/E)XK nuclease superfamily revisited: Detection of novel families with modified transitive meta profile searches. BMC Struct. Biol. 2007, 7, 40. [Google Scholar] [CrossRef] [PubMed]
- Steczkiewicz, K.; Muszewska, A.; Knizewski, L.; Rychlewski, L.; Ginalski, K. Sequence, structure and functional diversity of PD-(D/E)XK phosphodiesterase superfamily. Nucleic Acids Res. 2012, 40, 7016–7045. [Google Scholar] [CrossRef] [PubMed]
- Pingoud, A.; Fuxreiter, M.; Pingoud, V.; Wende, W. Type II restriction endonucleases: Structure and mechanism. Cell Mol. Life Sci. 2005, 62, 685–707. [Google Scholar] [CrossRef]
- Feder, M.; Bujnicki, J.M. Identification of a new family of putative PD-(D/E)XK nucleases with unusual phylogenomic distribution and a new type of the active site. BMC Genom. 2005, 6, 21. [Google Scholar] [CrossRef]
- Zhou, X.E.; Wang, Y.; Reuter, M.; Mucke, M.; Kruger, D.H.; Meehan, E.J.; Chen, L. Crystal structure of type IIE restriction endonuclease EcoRII reveals an autoinhibition mechanism by a novel effector-binding fold. J. Mol. Biol. 2004, 335, 307–319. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Park, J.H.; Inouye, M. Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 2011, 45, 61–79. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Inouye, M. mRNA interferases, sequence-specific endoribonucleases from the toxin-antitoxin systems. Prog. Mol. Biol. Transl. Sci. 2009, 85, 467–500. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Hoeflich, K.P.; Ikura, M.; Qing, G.; Inouye, M. MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli. Mol. Cell 2003, 12, 913–923. [Google Scholar] [CrossRef]
- Zhang, Y.; Inouye, M. The inhibitory mechanism of protein synthesis by YoeB, an Escherichia coli toxin. J. Biol. Chem. 2009, 284, 6627–6638. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yamaguchi, Y.; Inouye, M. Characterization of YafO, an Escherichia coli toxin. J. Biol. Chem. 2009, 284, 25522–25531. [Google Scholar] [CrossRef] [PubMed]
- Tram, U.; Ferree, P.M.; Sullivan, W. Identification of Wolbachia–host interacting factors through cytological analysis. Microb. Infect 2003, 5, 999–1011. [Google Scholar] [CrossRef]
- Bian, G.; Joshi, D.; Dong, Y.; Lu, P.; Zhou, G.; Pan, X.; Xu, Y.; Dimopoulos, G.; Xi, Z. Wolbachia invades Anopheles stephensi populations and induces refractoriness to Plasmodium infection. Science 2013, 340, 748–751. [Google Scholar] [CrossRef]
- Boyle, L.; O’Neill, S.L.; Robertson, H.M.; Karr, T.L. Interspecific and intraspecific horizontal transfer of Wolbachia in Drosophila. Science 1993, 260, 1796–1799. [Google Scholar] [CrossRef]
- Ye, Y.H.; Carrasco, A.M.; Frentiu, F.D.; Chenoweth, S.F.; Beebe, N.W.; van den Hurk, A.F.; Simmons, C.P.; O’Neill, S.L.; McGraw, E.A. Wolbachia Reduces the Transmission Potential of Dengue-Infected Aedes aegypti. PLoS Negl. Trop. Dis. 2015, 9, e0003894. [Google Scholar] [CrossRef]
- Blagrove, M.S.; Arias-Goeta, C.; Failloux, A.B.; Sinkins, S.P. Wolbachia strain wMel induces cytoplasmic incompatibility and blocks dengue transmission in Aedes albopictus. Proc. Natl. Acad. Sci. USA 2012, 109, 255–260. [Google Scholar] [CrossRef]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef]
- Pissaridou, P.; Allsopp, L.P.; Wettstadt, S.; Howard, S.A.; Mavridou, D.A.I.; Filloux, A. The Pseudomonas aeruginosa T6SS-VgrG1b spike is topped by a PAAR protein eliciting DNA damage to bacterial competitors. Proc. Natl. Acad. Sci. USA 2018, 115, 12519–12524. [Google Scholar] [CrossRef]
- Lara-Tejero, M.; Galan, J.E. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science 2000, 290, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Casabona, M.G.; Kneuper, H.; Chalmers, J.D.; Palmer, T. The type VII secretion system of Staphylococcus aureus secretes a nuclease toxin that targets competitor bacteria. Nat. Microbiol. 2016, 2, 16183. [Google Scholar] [CrossRef] [PubMed]
- Mercot, H.; Llorente, B.; Jacques, M.; Atlan, A.; Montchamp-Moreau, C. Variability within the Seychelles cytoplasmic incompatibility system in Drosophila simulans. Genetics 1995, 141, 1015–1023. [Google Scholar] [PubMed]
- Poinsot, D.; Bourtzis, K.; Markakis, G.; Savakis, C.; Mercot, H. Wolbachia transfer from Drosophila melanogaster into D. simulans: Host effect and cytoplasmic incompatibility relationships. Genetics 1998, 150, 227–237. [Google Scholar] [PubMed]
- Poinsot, D.; Charlat, S.; Mercot, H. On the mechanism of Wolbachia-induced cytoplasmic incompatibility: Confronting the models with the facts. Bioessays 2003, 25, 259–265. [Google Scholar] [CrossRef]
- Rao, P.N.; Johnson, R.T. Premature Chromosome Condensation: A Mechanism for the Elimination of Chromosomes in Virus-Fused Cells. J. Cell Sci. 1972, 10, 495–513. [Google Scholar]
- Ferree, P.M.; Sullivan, W. A genetic test of the role of the maternal pronucleus in Wolbachia-induced cytoplasmic incompatibility in Drosophila melanogaster. Genetics 2006, 173, 839–847. [Google Scholar] [CrossRef]
- Karr, T.L.; Kose, H.; Lassy, C. A ‘sink’ model for cytoplasmic incompatibility in Drosophila simulans: Interactions between host chromosomal proteins and Wolbachia in the egg and testis of Drosophila. Mol. Biol. Cell 1995, 6, 430a. [Google Scholar]
- Breeuwer, J.A.J.; Werren, J.H. Microorganisms associated with chromosome destruction and reproductive isolation between two insect species. Nature 1990, 346, 558–560. [Google Scholar] [CrossRef]
- Hurst, L.D. The evolution of cytoplasmic incompatibility or when spite can be successful. J. Theor. Biol. 1991, 148, 269–277. [Google Scholar] [CrossRef]
- Poinsot, D.; Mercot, H. Wolbachia can rescue from cytoplasmic incompatibility while being unable to induce it. In “From Symbiosis to Eukaryotism”: Endocytobiology VII: Proceedings of the International Congress on Endocytobiology, Symbiosis and Biomedicine; Wagner, E., Normann, J., Greppin, H., Hackstein, J.H.P., Herrmann, R.G., Kowallik, K.V., Schenk, H.E.A., Seckbach, J., Eds.; University of Geneva: Geneva, Switzerland, 1999; pp. 221–234. [Google Scholar]
- Shropshire, J.D.; Leigh, B.; Bordenstein, S.R.; Duplouy, A.; Riegler, M.; Brownlie, J.C.; Bordenstein, S.R. Models and Nomenclature for Cytoplasmic Incompatibility: Caution over Premature Conclusions—A Response to Beckmann et al. Trends Genet. 2019, 35, 397–399. [Google Scholar] [CrossRef]
- Bossan, B.; Koehncke, A.; Hammerstein, P. A new model and method for understanding Wolbachia-induced cytoplasmic incompatibility. PLoS ONE 2011, 6, e19757. [Google Scholar] [CrossRef] [PubMed]
- Coyne, E.S.; Wing, S.S. The business of deubiquitination-location, location, location. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Brohi, R.D.; Huo, L.J. Posttranslational Modifications in Spermatozoa and Effects on Male Fertility and Sperm Viability. OMICS 2017, 21, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, J.F.; Bonneau, M.; Chen, H.; Hochstrasser, M.; Poinsot, D.; Mercot, H.; Weill, M.; Sicard, M.; Charlat, S. Caution Does Not Preclude Predictive and Testable Models of Cytoplasmic Incompatibility: A Reply to Shropshire et al. Trends Genet. 2019, 35, 399–400. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.K.; Mikkelsen, M.; Pedersen, K.; Gerdes, K. RelE, a global inhibitor of translation, is activated during nutritional stress. Proc. Natl. Acad. Sci. USA 2001, 98, 14328–14333. [Google Scholar] [CrossRef]
- Christensen, S.K.; Pedersen, K.; Hansen, F.G.; Gerdes, K. Toxin-antitoxin loci as stress-response-elements: ChpAK/MazF and ChpBK cleave translated RNAs and are counteracted by tmRNA. J. Mol. Biol. 2003, 332, 809–819. [Google Scholar] [CrossRef]
- Hoffmann, A.A.; Ross, P.A.; Rasic, G. Wolbachia strains for disease control: Ecological and evolutionary considerations. Evol. Appl. 2015, 8, 751–768. [Google Scholar] [CrossRef]
- Ross, P.A.; Turelli, M.; Hoffmann, A.A. Evolutionary Ecology of Wolbachia Releases for Disease Control. Annu. Rev. Genet. 2019, 53, 93–116. [Google Scholar] [CrossRef]
- Biwot, J.C.; Zhang, H.B.; Liu, C.; Qiao, J.X.; Yu, X.Q.; Wang, Y.F. Wolbachia-induced expression of kenny gene in testes affects male fertility in Drosophila melanogaster. Insect Sci. 2019. [Google Scholar] [CrossRef]
- Israel, A. The IKK complex, a central regulator of NF-kB activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Shen, W.; Bi, J.; Chen, M.Y.; Wang, R.F.; Ai, H.; Wang, Y.F. Small RNA analysis provides new insights into cytoplasmic incompatibility in Drosophila melanogaster induced by Wolbachia. J. Insect Physiol. 2019, 118, 103938. [Google Scholar] [CrossRef] [PubMed]
- Harumoto, T.; Lemaitre, B. Male-killing toxin in a bacterial symbiont of Drosophila. Nature 2018, 557, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, M.D.; Gnirke, A.; Margolis, J.; Garnes, J.; Campbell, M.; Stuart, J.J.; Aggarwal, R.; Richards, S.; Park, Y.; Beeman, R.W. The maternal-effect, selfish genetic element Medea is associated with a composite Tc1 transposon. Proc. Natl. Acad. Sci. USA 2008, 105, 10085–10089. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Model | Mode of CI | Mode of Rescue | Reference | |
|---|---|---|---|---|
| Early models (pre-Cifs) | Mis-timing (“Slow-motion”) | Wolbachia infection slows down male pronuclear development. The resultant asynchrony of the parental and maternal pronucleus development causes CI. | Maternal pronucleus in Wolbachia-infected egg experiences a similar delay, or Wolbachia-infected egg accelerates male pronuclear development, resulting in developmental synchronization with the delayed male pronucleus. | [14,31,32,95,103] |
| Titration–Restitution (“Sink”) | A key host component involved in proper chromosome condensation and segregation is removed from male chromosomes by a Wolbachia effector. | Wolbachia-infected egg replenishes such components to the male chromosomes. | [23,95,98] | |
| “Lock and Key” | During spermatogenesis, Wolbachia deposit a “lock” factor that binds to certain component(s) of the paternal nucleus, interrupting its normal function. | Wolbachia-infected egg produces a “key” that specifically interacts with the “lock” and removes it from the paternal material. | [23,99,100,101] | |
| Models proposed after the discovery of Cifs | Toxin-antidote | Wolbachia secrete both CI toxin and antidote (CifB and CifA) during spermatogenesis. The rapid degradation of antidote protein in the mature sperm or fertilized egg activates the toxin which causes chromosomal defects in the male pronucleus. | Antidote protein (CifA) provided by Wolbachia-infected egg rescues the defect caused by the toxin, likely through direct binding to the toxin. | [24,38,100] |
| Two-by-one | A pair of Wolbachia effectors CifA and CifB function together to cause CI. CifA and CifB can induce CI either in a “toxin-antidote” mode, similar to described above; or in a “host modification” (HM) mode, where they induce CI by delaying the paternal pronucleus or modifying testis-specific host factor(s). | CifA protein produced in the infected egg rescues CI by acting as an antidote or modifying certain host factors in the embryo. | [41] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Zhang, M.; Hochstrasser, M. The Biochemistry of Cytoplasmic Incompatibility Caused by Endosymbiotic Bacteria. Genes 2020, 11, 852. https://doi.org/10.3390/genes11080852
Chen H, Zhang M, Hochstrasser M. The Biochemistry of Cytoplasmic Incompatibility Caused by Endosymbiotic Bacteria. Genes. 2020; 11(8):852. https://doi.org/10.3390/genes11080852
Chicago/Turabian StyleChen, Hongli, Mengwen Zhang, and Mark Hochstrasser. 2020. "The Biochemistry of Cytoplasmic Incompatibility Caused by Endosymbiotic Bacteria" Genes 11, no. 8: 852. https://doi.org/10.3390/genes11080852
APA StyleChen, H., Zhang, M., & Hochstrasser, M. (2020). The Biochemistry of Cytoplasmic Incompatibility Caused by Endosymbiotic Bacteria. Genes, 11(8), 852. https://doi.org/10.3390/genes11080852