Genetic Basis of Maize Resistance to Multiple Insect Pests: Integrated Genome-Wide Comparative Mapping and Candidate Gene Prioritization

 , ,
, ,  , ,
, , 

Abstract
1. Introduction
2. Material and Methods
2.1. Association Mapping Panel (AMP) Establishment and Field Planting
2.2. Genotyping and Quality Control and Assurance of SNP Markers
2.3. FAW Damage Scoring and MW Bioassay
2.4. Statistical Analyses of the Phenotypic Data
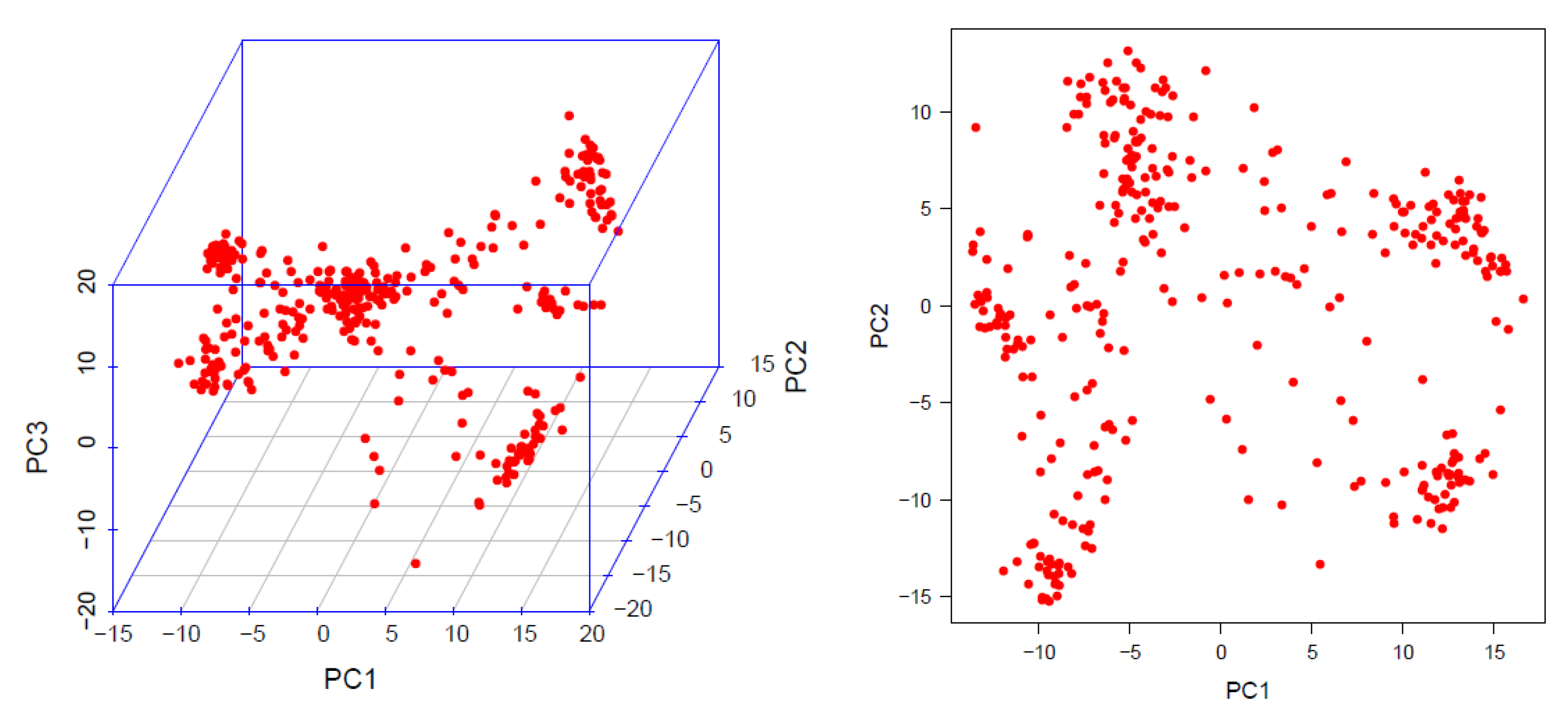
2.5. Linkage Disequilibrium (LD), Population Structure and Kinship Matrix
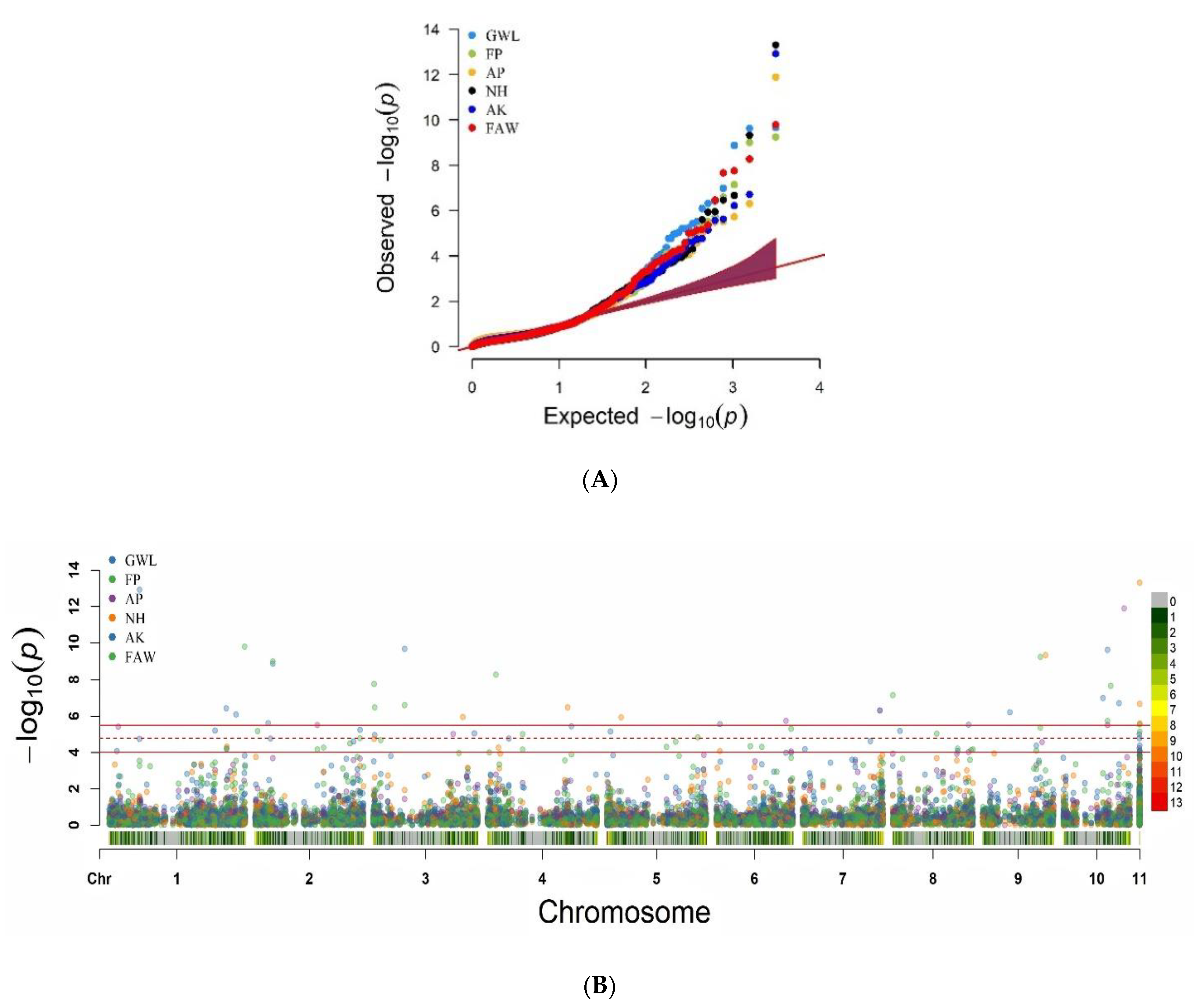
2.6. Genome-Wide Association Mapping
3. Candidate Gene (CG) Designation
Pre-CGs (Pre-CGs) Identification around the QTNs for Maize Resistance to FAW and MW
4. Pre-CGs Prioritization Through a Suite of Functional Characterizations
4.1. In-Silico Expression Analyses of the Pre-CGs
4.2. Identification of Conserved Domains within the Protein Sequences of the Pre-CGs
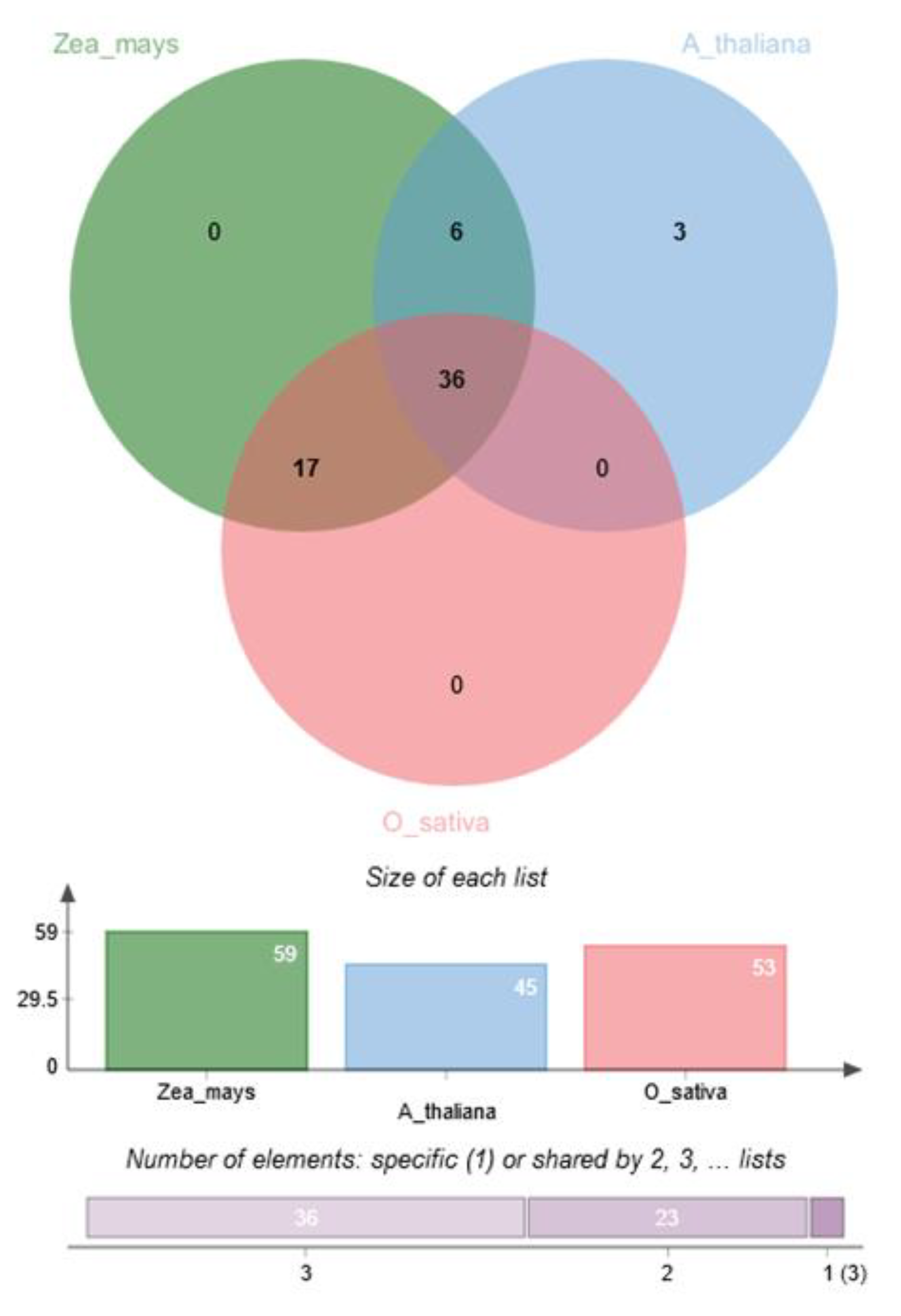
4.3. Identification of Pre-CG Orthologs and Co-Expression Analysis
4.4. Pre-CG Prioritization
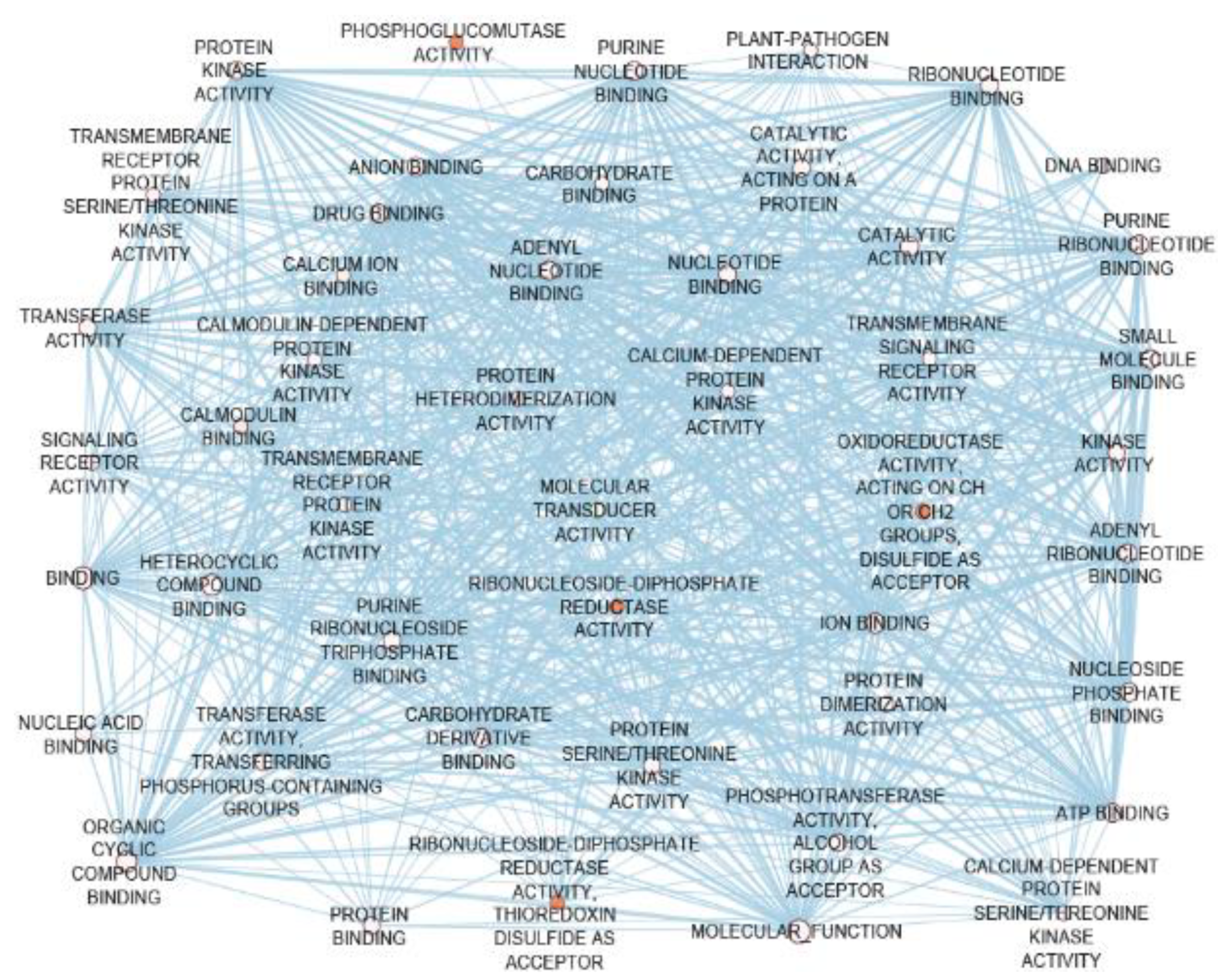
4.5. Network-Assisted CG Discovery for Multiple-Insect Resistance
4.6. Interactions Among CGs
5. Results
5.1. Trait Variance, Heritability, and Correlations
5.2. Association Mapping for MW and FAW Resistance Traits
5.3. Resistance-Related QTN-QTL-MQTL Co-Localizations
5.4. Pre-CGs Functionally Associated with Plant Stress Response Mechanisms in the Vicinity of the QTNs
5.5. Pre-CGs Differentially Expressed under Biotic and Abiotic Stress Conditions
5.6. Pre-CGs were Co-Expressed with Their Rice and Arabidopsis Thaliana Ortholog Genes
5.7. NbCGs were Biologically Connected to the GbCGs
6. Discussion and Conclusions
6.1. Association Mapping Panel
6.2. Linkage Disequilibrium and Control of False-Positive and Negative Association
6.3. QTNs for Both Single and Combined Maize Resistance to FAW and MW
6.4. Resistance Across Insect Pest Species and Maize Organs
6.5. Promising CGs for Maize Resistance to Multiple-Insect Pests
6.6. Research and Breeding Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tefera, T.; Goftishu, M.; Ba, M.; Rangaswamy, M. A Guide to Biological Control of Fall Armyworm in Africa Using Egg Parasitoids, 1st ed.; International Centre of Insect Physiology and Ecology: Nairobi, Kenya, 2019. [Google Scholar]
- Renzaho, A.M.N.; Kamara, J.K.; Toole, M. Biofuel production and its impact on food security in low and middle income countries: Implications for the post-2015 sustainable development goals. Renew. Sustain. Energy Rev. 2017, 78, 503–516. [Google Scholar] [CrossRef]
- James, A.; Zikankuba, V.L. Mycotoxins contamination in maize alarms food safety in sub-Sahara Africa. Food Control 2018, 90, 372–381. [Google Scholar] [CrossRef]
- Meihls, L.N.; Kaur, H.; Jander, G. Natural Variation in Maize Defense against Insect Herbivores. Cold Spring Harb. Symp. Quant. Biol. 2012, 77, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Midega, C.A.O.; Murage, A.W.; Pittchar, J.O.; Khan, Z.R. Managing storage pests of maize: Farmers’ knowledge, perceptions and practices in western Kenya. Crop Prot. 2016, 90, 142–149. [Google Scholar] [CrossRef]
- Nyukuri, R.W.; Wanjala, F.M.; Kirui, S.C.; Cheramgoi, E.; Chirchir, E.; Mwale, R. Amage of stem borer species to zea mays l., sorghum bicolor l.and three refugia graminae. Adv. Agric. Biol. 2014, 1, 37–45. [Google Scholar]
- Goergen, G.; Kumar, P.L.; Sankung, S.B.; Togola, A.; Tamò, M. First report of outbreaks of the fall armyworm spodoptera frugiperda (J E Smith) (Lepidoptera, Noctuidae), a new alien invasive pest in West and Central Africa. PLoS ONE 2016, 11, e0165632. [Google Scholar] [CrossRef]
- Padhee, A.K.; Prassanna, B.M. The emerging threat of Fall Armyworm in India. Indian Farming 2019, 69, 51–54. [Google Scholar]
- Kumar, D.; Kalita, P. Reducing Postharvest Losses during Storage of Grain Crops to Strengthen Food Security in Developing Countries. Foods 2017, 6, 8. [Google Scholar] [CrossRef]
- Kebede, M.; Shimalis, T. Out-break, Distribution and Management of fall armyworm, Spodoptera frugiperda J.E. Smith in Africa: The Status and Prospects. Acad. Agric. J. 2018, 3, 551–568. [Google Scholar]
- Devi, S. Fall armyworm threatens food security in southern Africa. Lancet 2018, 391, 727. [Google Scholar] [CrossRef]
- García-lara, S.; Burt, A.J.; Arnason, J.T.; Bergvinson, D.J. QTL Mapping of Tropical Maize Grain Components Associated with Maize Weevil Resistance. Crop Sci. 2010, 50, 815–825. [Google Scholar] [CrossRef]
- Murenga, M.; Derera, J.; Mugo, S.; Tongoona, P. A review of genetic analysis and response to selection for resistance to Busseola fusca and Chilo partellus, stem borers in tropical maize germplasm: A Kenyan perspective. Maydica 2016, 61, M4. [Google Scholar]
- Mihm, J.A. Maize pest management—multiple insect and disease resistant varieties are the key to success. In Proceedings of the Fourth Eastern and Southern Africa Regional Maize Conference, Harare, Zimbabwe, 28 March–1 April 1994; Jewel, D.C., Waddington, S.R., Ransom, J.K., Pixely, K.V., Eds.; International Maize and Wheat Improvement Center (CIMMYT): El Batan, Mexico, 1995; pp. 176–181. [Google Scholar]
- Thoen, M.P.M.; Olivas, N.D.; Kloth, K.J.; Coolen, S.; Huang, P.-P.; Aarts, M.G.M.; Bac-Molenaar, J.A.; Bakker, J.; Bouwmeester, H.J.; Broekgaarden, C.; et al. Genetic architecture of plant stress resistance: Multi-trait genome-wide association mapping. New Phytol. 2016, 213, 1346–1362. [Google Scholar] [CrossRef] [PubMed]
- Badji, A.; Otim, M.; Machida, L.; Odong, T.; Kwemoi, D.B.; Okii, D.; Agbahoungba, S.; Mwila, N.; Kumi, F.; Ibanda, A.; et al. Maize Combined Insect Resistance Genomic Regions and Their Co-localization With Cell Wall Constituents Revealed by Tissue-Specific QTL Meta-Analyses. Front. Plant Sci. 2018, 9, 895. [Google Scholar] [CrossRef]
- Munyiri, S.W.; Mugo, S.N. Quantitative trait loci for resistance to spotted and African maize stem borers (Chilo partellus and Busseola fusca) in a tropical maize (Zea mays L.) population. Afr. J. Biotechnol. 2017, 16, 1579–1589. [Google Scholar] [CrossRef]
- Mwololo, J.K. Resistance in Tropical Maize To the Maize Weevil and Larger Grain Borer. Ph.D. Thesis, Makerere University, Kampala, Uganda, 2013. [Google Scholar]
- War, A.R.; Paulraj, M.G.; Ahmad, T.; Buhroo, A.A.; Hussain, B.; Ignacimuthu, S.; Sharma, H.C. Mechanisms of plant defense against insect herbivores. Plant Signal. Behav. 2012, 7, 1306–1320. [Google Scholar] [CrossRef]
- Kliebenstein, D.J. Quantitative Genetics and Genomics of Plant Resistance to Insects. Annu. Plant Rev. 2014, 47, 235–262. [Google Scholar] [CrossRef]
- Barah, P.; Bones, A.M. Multidimensional approaches for studying plant defence against insects: From ecology to omics and synthetic biology. J. Exp. Bot. 2015, 66, 479–493. [Google Scholar] [CrossRef]
- Chakradhar, T.; Hindu, V.; Reddy, P.S. Genomic-based-breeding tools for tropical maize improvement. Genetica 2017, 145, 525–539. [Google Scholar] [CrossRef]
- Ishikawa, A. A strategy for identifying quantitative trait genes using gene expression analysis and causal analysis. Genes 2017, 8, 347. [Google Scholar] [CrossRef]
- Sitonik, C.; Suresh, L.M.; Beyene, Y.; Olsen, M.S.; Makumbi, D.; Oliver, K.; Das, B.; Bright, J.M.; Mugo, S.; Crossa, J.; et al. Genetic architecture of maize chlorotic mottle virus and maize lethal necrosis through GWAS, linkage analysis and genomic prediction in tropical maize germplasm. Theor. Appl. Genet. 2019, 132, 2381–2399. [Google Scholar] [CrossRef] [PubMed]
- Samayoa, L.F.; Malvar, R.A.; Olukolu, B.A.; Holland, J.B.; Butrón, A. Genome-wide association study reveals a set of genes associated with resistance to the Mediterranean corn borer (Sesamia nonagrioides L.) in a maize diversity panel. BMC Plant Biol. 2015, 15, 35. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Galindo, J.C.; Malvar, R.A.; Butrón, A.; Santiago, R.; Samayoa, L.F.; Caicedo, M.; Ordás, B. Mapping of resistance to corn borers in a MAGIC population of maize. BMC Plant Boil. 2019, 19, 431. [Google Scholar] [CrossRef] [PubMed]
- Nyaga, C.; Gowda, M.; Beyene, Y.; Murithi, W.T.; Makumbi, D.; Olsen, M.S.; Suresh, M.L.; Bright, J.M.; Das, B.; Prasanna, B.M. Genome-Wide Analyses and Prediction of Resistance to MLN in Large Tropical Maize Germplasm. Genes 2019, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Jia, P.; Zhao, Z. Network-assisted analysis to prioritize GWAS results: Principles, methods and perspectives. Hum. Genet. 2014, 133, 125–138. [Google Scholar] [CrossRef]
- Zhang, Y.; Jia, Z.; Dunwell, J.M. Editorial: The Applications of New Multi-Locus GWAS Methodologies in the Genetic Dissection of Complex Traits. Front. Plant Sci. 2019, 10, 100. [Google Scholar] [CrossRef]
- Burghardt, L.T.; Young, N.D.; Tiffin, P. A Guide to Genome-Wide Association Mapping in Plants. Curr. Protoc. Plant Biol. 2017, 2, 22–38. [Google Scholar] [CrossRef]
- Schaefer, R.; Michno, J.-M.; Jeffers, J.; Hoekenga, O.A.; Dilkes, B.P.; Baxter, I.; Myers, C.L. Integrating Coexpression Networks with GWAS to Prioritize Causal Genes in Maize. Plant Cell 2018, 30, 2922–2942. [Google Scholar] [CrossRef]
- Andorf, C.M.; Cannon, E.K.S.; Portwood, J.L.; Gardiner, J.M.; Harper, L.C.; Schaeffer, M.L.; Braun, B.L.; Campbell, D.; Vinnakota, A.G.; Sribalusu, V.V.; et al. MaizeGDB update: New tools, data and interface for the maize model organism database. Nucleic Acids Res. 2015, 44, D1195–D1201. [Google Scholar] [CrossRef]
- Muthuramalingam, P.; Krishnan, S.R.; Pothiraj, R. Global Transcriptome Analysis of Combined Abiotic Stress Signaling Genes Unravels Key Players in Oryza sativa L.: An In silico Approach. Front. Plant Sci. 2017, 8, 759. [Google Scholar] [CrossRef]
- Woldesemayat, A.A.; Modise, D.M.; Gemeildien, J.; Ndimba, B.K.; Christoffels, A. Cross-species multiple environmental stress responses: An integrated approach to identify candidate genes for multiple stress tolerance in sorghum (Sorghum bicolor (L.) Moench) and related model species. PLoS ONE 2018, 13, e0192678. [Google Scholar]
- Akhunov, E.; Sehgal, S.; Liang, H.; Wang, S.; Akhunova, A.R.; Kaur, G.; Li, W.; Forrest, K.L.; See, D.; Šimková, H.; et al. Comparative analysis of syntenic genes in grass genomes reveals accelerated rates of gene structure and coding sequence evolution in polyploid wheat. Plant Physiol. 2012, 161, 252–265. [Google Scholar] [CrossRef]
- Gabaldón, T.; Koonin, E.V. Functional and evolutionary implications of gene orthology. Nat. Rev. Genet. 2015, 91, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Liu, T.; Li, B.; Sui, Y.; Chen, J.; Shi, J.; Wing, R.A.; Chen, M. Comparative Sequence Analysis of the Ghd7 Orthologous Regions Revealed Movement of Ghd7 in the Grass Genomes. PLoS ONE 2012, 7, e50236. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Shah, T.; Hao, Z.; Taba, S.; Zhang, S.; Gao, S.; Liu, J.; Cao, M.; Wang, J.; Prakash, A.B.; et al. Comparative SNP and Haplotype Analysis Reveals a Higher Genetic Diversity and Rapider LD Decay in Tropical than Temperate Germplasm in Maize. PLoS ONE 2011, 6, e24861. [Google Scholar] [CrossRef] [PubMed]
- Munyiri, W.S.; Mugo, N.S.; Otim, M.; Tefera, T.; Beyene, Y.; Mwololo, K.J.; Okori, P. Responses of tropical maize landraces to damage by Chilo partellus stem borer. Afr. J. Biotechnol. 2013, 12, 1229–1235. [Google Scholar]
- Munyiri, S.W.; Mugo, S.N.; Otim, M.; Mwololo, J.K.; Okori, P. Mechanisms and Sources of Resistance in Tropical Maize Inbred Lines to Chilo partellus Stem Borers. J. Agric. Sci. 2013, 5, 51–60. [Google Scholar] [CrossRef][Green Version]
- Mwololo, J.K.; Mugo, S.; Okori, P.; Tefera, T.; Otim, M.; Munyiri, S.W. Sources of Resistance to the Maize Weevil Sitophilus Zeamais in Tropical Maize. J. Agric. Sci. 2012, 4, 206–215. [Google Scholar]
- Mwololo, J.; Okori, P.; Mugo, S.; Tefera, T.; Yoseph, B.; Otim, M.; Munyiri, S.W. Phenotypic and Genotypic Variation in Tropical Maize Inbred Lines for Resistance To the Maize Weevil and Larger Grain Borer. Int. J. Agric. Sci. Res. 2012, 2, 41–52. [Google Scholar]
- Dramadri, I.O.; Nkalubo, S.T.; Kelly, J.D. Identification of QTL Associated with Drought Tolerance in Andean Common Bean. Crop Sci. 2019, 59, 1007–1020. [Google Scholar] [CrossRef]
- Sansaloni, C.; Petroli, C.; Jaccoud, D.; Carling, J.; Detering, F.; Grattapaglia, D.; Kilian, A. Diversity Arrays Technology (DArT) and next-generation sequencing combined: Genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proc. 2011, 5 (Suppl. S7), 54. [Google Scholar] [CrossRef]
- Gruber, B.; Unmack, P.J.; Berry, O.F.; Georges, A. dartr: An r package to facilitate analysis of SNP data generated from reduced representation genome sequencing. Mol. Ecol. Resour. 2018, 18, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Davis, F.M.; Ng, S.S.; Williams, W.P. Visual rating scales for screening whorl-stage corn for resistance to fall armyworm. Tech. Bull. Agric. For. Exp. Stn. 1992, 186, 1–9. [Google Scholar]
- Prasanna, B.M.; Huesing, J.E.; Eddy, R.; Peschke, V.M.; Regina, E.; Virginia, M.P. Fall Armyworm in Africa: A guide for integrated pest management, 1st ed. In Proceedings of the West Africa Regional Training of Trainers and Awareness Generation Workshop on Fall Armyworm Management, IITA, Cotonou, Bénin, 13–16 February 2018; Prasanna, B.M., Regina, E., Virginia, M.P., Eds.; CIMMYT: El Batan, Mexico, 2018. [Google Scholar]
- Sodedji, F.A.K.; Kwemoi, D.B.; Asea, G.; Kyamanywa, S. Response of provitamin-A maize germplasm to storage weevil Sitophilus zeamais (Motschulsky). Int. J. Agron. Agric. Res. 2016, 9, 1–13. [Google Scholar]
- Kasozi, L.C.; Derera, J.; Tongoona, P.; Zziwa, S.; Muwonge, A.; Gasura, E.; Bergvinson, D.J. Comparing the Effectiveness of the “ weevil warehouse ” and “ laboratory bioassay ” as Techniques for Screening Maize Genotypes for Weevil Resistance. J. Food Secur. 2018, 6, 170–177. [Google Scholar]
- Bates, D.M.; Maechler, M.; Bolker, B.; Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- R Development Core Team R. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2011; Volume 1, p. 409. [Google Scholar]
- Holland, J.B.; Nyquist, W.E.; Cervantes-Martinez, C.T. Estimating and Interpreting Heritability for Plant Breeding: An Update. Plant Breed. Rev. 2003, 22, 9–112. [Google Scholar]
- Marroni, F.; Pinosio, S.; Zaina, G.; Fogolari, F.; Felice, N.; Cattonaro, F.; Morgante, M. Nucleotide diversity and linkage disequilibrium in Populus nigra cinnamyl alcohol dehydrogenase (CAD4) gene. Tree Genet. Genomes 2011, 7, 1011–1023. [Google Scholar] [CrossRef]
- Remington, D.L.; Thornsberry, J.M.; Matsuoka, Y.; Wilson, L.M.; Whitt, S.R.; Doebley, J.; Kresovich, S.; Goodman, M.M.; Buckler, E.S. Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. USA 2001, 98, 11479–11484. [Google Scholar] [CrossRef]
- Peterson, R.A. Package ‘bestNormalize’. 2018. Available online: https://CRAN.R-project.org/package=bestNormalize (accessed on 25 April 2020).
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, X.; Wang, J.; Li, M.; Wang, Q.; Tian, F.; Su, Z.; Pan, Y.; Liu, D.; Lipka, A.E.; et al. GAPIT Version 2: An Enhanced Integrated Tool for Genomic Association and Prediction. Plant Genome 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-M.; Shao, X.-Y.; Pei, Y.-H.; Guo, X.-M.; Li, J.; Song, X.-Y.; Zhao, M.-A. Genetic Diversity and Genome-Wide Association Study of Major Ear Quantitative Traits Using High-Density SNPs in Maize. Front. Plant Sci. 2018, 9, 966. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.M.; Mao, Y.; Xie, C.; Smith, H.; Luo, L.; Xu, S. Mapping quantitative trait loci using naturally occurring genetic variance among commercial inbred lines of maize (Zea mays L.). Genetics 2005, 169, 2267–2275. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Tucker, M.M.; Basten, C.J.; Gandhi, H.; Ersoz, E.; Guo, B.; Xu, Z.; Wang, D.; Gay, G. The impact of population structure on genomic prediction in stratified populations. Theor. Appl. Genet. 2014, 127, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yu, Y.; Yuan, J.; Zhang, X.; Huang, H.; Li, F.; Xiang, J. Effects of marker density and population structure on the genomic prediction accuracy for growth trait in Pacific white shrimp Litopenaeus vannamei. BMC Genet. 2017, 18, 45. [Google Scholar] [CrossRef]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef]
- Li, X.; He, K.; Wang, Z.; Bai, S. Quantitative Trait Loci for Asian Corn Borer Resistance in Maize Population Mc37 × Zi330. Agric. Sci. China 2010, 9, 77–84. [Google Scholar] [CrossRef]
- Womack, E.D.; Warburton, M.L.; Williams, W.P. Mapping of quantitative trait loci for resistance to fall armyworm and southwestern corn borer leaf-feeding damage in maize. Crop Sci. 2018, 58, 529–539. [Google Scholar] [CrossRef]
- Brooks, T.D.; Willcox, M.C.; Williams, W.P.; Buckley, P.M. Quantitative trait loci conferring resistance to fall armyworm and southwestern corn borer leaf feeding damage. Crop Sci. 2005, 45, 2430–2434. [Google Scholar] [CrossRef]
- Brooks, T.D.; Bushman, B.S.; Williams, W.P.; McMullen, M.D.; Buckley, P.M. Genetic basis of resistance to fall armyworm (Lepidoptera: Noctuidae) and southwestern corn borer (Lepidoptera: Crambidae) leaf-feeding damage in maize. J. Econ. Entomol. 2007, 100, 1470–1475. [Google Scholar] [CrossRef] [PubMed]
- Butrón, A.; Chen, Y.C.; Rottinghaus, G.E.; McMullen, M.D.; Butro, A.; McMullen, M.D. Genetic variation at bx1 controls DIMBOA content in maize. Theor. Appl. Genet. 2010, 120, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Meihls, L.N.; Handrick, V.; Glauser, G.; Barbier, H.; Kaur, H.; Haribal, M.M.; Lipka, A.E.; Gershenzon, J.; Buckler, E.S.; Erb, M.; et al. Natural Variation in Maize Aphid Resistance Is Associated with 2,4-Dihydroxy-7-Methoxy-1,4-Benzoxazin-3-One Glucoside Methyltransferase Activity[C][W]. Plant Cell 2013, 25, 2341–2355. [Google Scholar] [CrossRef]
- Betsiashvili, M.; Ahern, K.R.; Jander, G. Additive effects of two quantitative trait loci that confer Rhopalosiphum maidis (corn leaf aphid) resistance in maize inbred line Mo17. J. Exp. Bot. 2015, 66, 571–578. [Google Scholar] [CrossRef]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93. [Google Scholar] [CrossRef]
- Hruz, T.; Wyss, M.; Docquier, M.; Pfaffl, M.W.; Masanetz, S.; Borghi, L.; Verbrugghe, P.; Kalaydjieva, L.; Bleuler, S.; Laule, O.; et al. RefGenes: Identification of reliable and condition specific reference genes for RT-qPCR data normalization. BMC Genom. 2011, 12, 156. [Google Scholar] [CrossRef]
- Petryszak, R.; Burdett, T.; Fiorelli, B.; Fonseca, N.A.; Gonzalez-Porta, M.; Hastings, E.; Huber, W.; Jupp, S.; Keays, M.; Kryvych, N.; et al. Expression Atlas update—A database of gene and transcript expression from microarray- and sequencing-based functional genomics experiments. Nucleic Acids Res. 2013, 42, D926–D932. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2016, 45, D200–D203. [Google Scholar] [CrossRef]
- Wang, Y.; Coleman-Derr, D.; Chen, G.; Gu, Y.Q. OrthoVenn: A web server for genome wide comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2015, 43, W78–W84. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Lee, S.; Yang, S.; Lee, I. MaizeNet: A co-functional network for network-assisted systems genetics in Zea mays. Plant J. 2019, 99, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2018, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Reimand, J.; Isserlin, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C.; et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef] [PubMed]
- Isserlin, R.; Merico, D.; Voisin, V.; Bader, G.D. Enrichment Map—A Cytoscape app to visualize and explore OMICs pathway enrichment results. F1000Research 2014, 3, 141. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Williams, W.P.; Mihm, J.A.; Barry, B.D.; Overman, J.L.; Wiseman, B.R.; Riley, T.J. Resistance to multiple lepidopterous species in tropical derived corn germplasm. Tech. Bull. Agric. For. Exp. Stn. 1988, 157, 8. [Google Scholar]
- Bolker, B.; Brooks, M.E.; Clark, C.J.; Geange, S.W.; Poulsen, J.R.; Stevens, M.H.H.; White, J.-S.S. Generalized linear mixed models: A practical guide for ecology and evolution. Trends Ecol. Evol. 2009, 24, 127–135. [Google Scholar] [CrossRef]
- Dhliwayo, T.; Pixley, K.V. Divergent selection for resistance to maize weevil in six maize populations. Crop Sci. 2003, 43, 2043–2049. [Google Scholar] [CrossRef]
- Otim, M.H.; Tay, W.T.; Walsh, T.K.; Kanyesigye, D.; Adumo, S.; Abongosi, J.; Ochen, S.; Sserumaga, J.P.; Alibu, S.; Abalo, G.; et al. Detection of sister-species in invasive populations of the fall armyworm Spodoptera frugiperda (Lepidoptera: Noctuidae) from Uganda. PLoS ONE 2018, 13, e0194571. [Google Scholar] [CrossRef]
- Arief, V.N.; Desmae, H.; Hardner, C.; DeLacy, I.H.; Gilmour, A.; Bull, J.K.; Basford, K.E. Utilization of Multi-year Plant Breeding Data to Get Better Prediction of Genotype Performance. Crop Sci. 2018, 59, 480–490. [Google Scholar] [CrossRef]
- Babic, M.; Andjelkovic, V.; Babic, V. Genotype by environment interaction in maize breeding. Genetika 2008, 40, 303–312. [Google Scholar] [CrossRef]
- Barros-Rios, J.; Malvar, R.A.; Jung, H.-J.G.; Santiago, R. Cell wall composition as a maize defense mechanism against corn borers. Phytochemistry 2011, 72, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Santiago, R.; Barros-Rios, J.; Malvar, R.A. Impact of cell wall composition on maize resistance to pests and diseases. Int. J. Mol. Sci. 2013, 14, 6960–6980. [Google Scholar] [CrossRef] [PubMed]
- García-Lara, S.; Bergvinson, D.J. Phytochemical and Nutraceutical Changes during Recurrent Selection for Storage Pest Resistance in Tropical Maize. Crop Sci. 2014, 54, 2423. [Google Scholar] [CrossRef]
- Zaidi, P.H.; Seetharam, K.; Krishna, G.; Krishnamurthy, L.; Gajanan, S.; Babu, R.; Zerka, M.; Vinayan, M.T.; Vivek, B.S. Genomic regions associated with root traits under drought stress in tropical maize (Zea mays L.). PLoS ONE 2016, 11, e0164340. [Google Scholar] [CrossRef] [PubMed]
- Romay, M.C.; Millard, M.J.; Glaubitz, J.C.; Peiffer, J.A.; Swarts, K.; Casstevens, T.; Elshire, R.; Acharya, C.B.; Mitchell, S.E.; Flint-Garcia, S.; et al. Comprehensive genotyping of the USA national maize inbred seed bank. Genome Biol. 2013, 14, R55. [Google Scholar] [CrossRef]
- Chaikam, V.; Gowda, M.; Nair, S.K.; Melchinger, A.E.; Boddupalli, P.M. Genome-wide association study to identify genomic regions influencing spontaneous fertility in maize haploids. Euphytica 2019, 215, 138. [Google Scholar] [CrossRef]
- Marees, A.T.; De Kluiver, H.; Stringer, S.; Vorspan, F.; Curis, E.; Marie-Claire, C.; Derks, E.M. A tutorial on conducting genome-wide association studies: Quality control and statistical analysis. Int. J. Methods Psychiatr. Res. 2018, 27, e1608. [Google Scholar] [CrossRef]
- García-Lara, S.; Khairallah, M.M.; Vargas, M.; Bergvinson, D.J. Mapping of QTL Associated with Maize Weevil Resistance in Tropical Maize. Crop Sci. 2009, 49, 139–149. [Google Scholar] [CrossRef]
- Castro-Álvarez, F.F.; William, M.; Bergvinson, D.J.; García-Lara, S. Genetic mapping of QTL for maize weevil resistance in a RIL population of tropical maize. Theor. Appl. Genet. 2015, 128, 411–419. [Google Scholar] [CrossRef]
- Schulthess, A.W.; Reif, J.C.; Ling, J.; Plieske, J.; Kollers, S.; Ebmeyer, E.; Korzun, V.; Argillier, O.; Stiewe, G.; Ganal, M.W.; et al. The roles of pleiotropy and close linkage as revealed by association mapping of yield and correlated traits of wheat (Triticum aestivum L.). J. Exp. Bot. 2017, 68, 4089–4101. [Google Scholar] [CrossRef]
- Mikić, S.; Ahmad, S. Benzoxazinoids-protective secondary metabolites in cereals: The role and application. Ratar. Povrt. 2018, 55, 49–57. [Google Scholar] [CrossRef]
- Niculaes, C.; Abramov, A.; Hannemann, L.; Frey, M. Plant Protection by Benzoxazinoids—Recent Insights into Biosynthesis and Function. Agronomy 2018, 8, 143. [Google Scholar] [CrossRef]
- Zhou, S.; Richter, A.; Jander, G. Beyond defense: Multiple functions of benzoxazinoids in maize metabolism. Plant Cell Physiol. 2018, 59, 1528–1537. [Google Scholar] [CrossRef]
- Hassani-Pak, K.; Rawlings, C. Knowledge Discovery in Biological Databases for Revealing Candidate Genes Linked to Complex Phenotypes. J. Integr. Bioinform. 2017, 14, 1–9. [Google Scholar] [CrossRef]
- Kemper, K.E.; Bowman, P.J.; Hayes, B.J.; Visscher, P.M.; Goddard, M.E. A multi-trait Bayesian method for mapping QTL and genomic prediction. Genet. Sel. Evol. 2018, 50, 1–13. [Google Scholar] [CrossRef]
- Xiang, R.; MacLeod, I.M.; Bolormaa, S.; Goddard, M.E. Genome-wide comparative analyses of correlated and uncorrelated phenotypes identify major pleiotropic variants in dairy cattle. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Paaby, A.B.; Rockman, M.V. The many faces of pleiotropy. Trends Genet. 2013, 29, 66–73. [Google Scholar] [CrossRef]
- Visscher, P.M.; Yang, J. A plethora of pleiotropy across complex traits. Nat. Genet. 2016, 48, 707–708. [Google Scholar] [CrossRef]
- Omony, J. Biological Network Inference: A Review of Methods and Assessment of Tools and Techniques. Annu. Res. Rev. Biol. 2014, 4, 577–601. [Google Scholar] [CrossRef]
- Lee, T.; Kim, H.; Lee, I. Network-assisted crop systems genetics: Network inference and integrative analysis. Curr. Opin. Plant Biol. 2015, 24, 61–70. [Google Scholar] [CrossRef]
- Kissoudis, C.; van de Wiel, C.; Visser, R.G.F.; van der Linden, G. Enhancing crop resilience to combined abiotic and biotic stress through the dissection of physiological and molecular crosstalk. Front. Plant Sci. 2014, 5, 207. [Google Scholar] [CrossRef]
- Suzuki, N. Hormone signaling pathways under stress combinations. Plant Signal. Behav. 2016, 11, e1247139. [Google Scholar] [CrossRef]
- Baillo, E.H.; Kimotho, R.N.; Zhang, Z.; Xu, P. Transcription factors associated with abiotic and biotic stress tolerance and their potential for crops improvement. Genes 2019, 10, 771. [Google Scholar] [CrossRef]
- Vats, S. Biotic and Abiotic Stress Tolerance in Plants; Springer: Cham, Switzerland, 2018; Volume 5, pp. 26–33. [Google Scholar]
- Pandey, P.; Irulappan, V.; Bagavathiannan, M.V.; Senthil-Kumar, M. Impact of Combined Abiotic and Biotic Stresses on Plant Growth and Avenues for Crop Improvement by Exploiting Physio-morphological Traits. Front. Plant Sci. 2017, 8, 178. [Google Scholar] [CrossRef]
- Nejat, N.; Mantri, N. Plant immune system: Crosstalk between responses to biotic and abiotic stresses the missing link in understanding plant defence. Curr. Issues Mol. Biol. 2017, 23, 1–16. [Google Scholar] [CrossRef]
- Butron, A.; Samayoa, L.F.; Santiago, R.; Ordás, B.; Malvar, R.A. Genomics of Insect Resistance. In The Maize Genome; Springer: Cham, Switzerland, 2018; pp. 163–183. [Google Scholar]
- López-Castillo, L.M.; Silva-Fernández, S.E.; Winkler, R.; Bergvinson, D.J.; Arnason, J.T.; García-Lara, S. Postharvest insect resistance in maize. J. Stored Prod. Res. 2018, 77, 66–76. [Google Scholar] [CrossRef]
- Ku, Y.S.; Sintaha, M.; Cheung, M.Y.; Lam, H.M. Plant hormone signaling crosstalks between biotic and abiotic stress responses. Int. J. Mol. Sci. 2018, 19, 3206. [Google Scholar] [CrossRef]
- Ramegowda, V.; Senthil-Kumar, M. The interactive effects of simultaneous biotic and abiotic stresses on plants: Mechanistic understanding from drought and pathogen combination. J. Plant Physiol. 2015, 176, 47–54. [Google Scholar] [CrossRef]
- Bruce, T.J.A. Interplay between insects and plants: Dynamic and complex interactions that have coevolved over millions of years but act in milliseconds. J. Exp. Bot. 2015, 66, 455–465. [Google Scholar] [CrossRef]
- Gimenez, E.; Salinas, M.; Manzano-Agugliaro, F. Worldwide research on plant defense against biotic stresses as improvement for sustainable agriculture. Sustainability 2018, 10, 391. [Google Scholar] [CrossRef]
- Glazebrook, J.; Roby, D. Plant biotic interactions: From conflict to collaboration. Plant J. 2018, 93, 589–591. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, M.E.; Arnaiz, A.; Gonzalez-Melendi, P.; Martinez, M.; Diaz, I. Plant perception and short-term responses to phytophagous insects and mites. Int. J. Mol. Sci. 2018, 19, 1356. [Google Scholar] [CrossRef]
- Gupta, P.K.; Kulwal, P.L.; Jaiswal, V. Association mapping in plants in the post-GWAS genomics era. In Advances in Genetics, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2019; pp. 1–80. [Google Scholar] [CrossRef]
- Andersen, E.J.; Ali, S.; Byamukama, E.; Yen, Y. Disease Resistance Mechanisms in Plants. Genes 2018, 9, 339. [Google Scholar] [CrossRef]
- Erb, M.; Reymond, P. Molecular Interactions Between Plants and Insect Herbivores. Annu. Rev. Plant Biol. 2019, 70, 527–557. [Google Scholar] [CrossRef]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of Total Genetic Value Using Genome-Wide Dense Marker Maps. Genetics 2001, 157, 1819–1829. [Google Scholar]
- Robertsen, C.D.; Hjortshøj, R.L.; Janss, L.L. Genomic Selection in Cereal Breeding. Agronomy 2019, 9, 95. [Google Scholar] [CrossRef]
- Liu, X.; Wang, H.; Wang, H.; Guo, Z.; Xu, X.; Liu, J.; Wang, S.; Prasanna, B.M.; Zou, C.; Prasanna, B.M.; et al. Factors affecting genomic selection revealed by empirical evidence in maize. Crop J. 2018, 6, 341–352. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SOURCE OF VARIATION | DF | 2017B | DF | 2019A | DF | 2018A | DF | ACROSS ENV. |
|---|---|---|---|---|---|---|---|---|
| GENOTYPE | 315 | 1.51 ** | 251 | 3.12 *** | 91 | 3.48 *** | 357 | 2.76 *** |
| BLOCK | 11 | 3.75 *** | 9 | 1.59 ns | 4 | 7.17 *** | 11 | 2.41 * |
| REPLICATION | 1 | 26.95 *** | ||||||
| ENVIRONMENT | 2 | 270.57 *** | ||||||
| GENOTYPE*ENVIRONMENT | 300 | 2.14 *** | ||||||
| RESIDUALS | 9 | 0.25 | 49 | 0.90 | 123 | 1.12 | 195 | 1.12 |
| H2 | 0.80 | 0.72 | 0.67 | 0.25 |
| SOURCE OF VARIATION | DF | AK | AP | FP | GWL | NH |
|---|---|---|---|---|---|---|
| GENOTYPE | 131 | 4795.47 *** | 5947.91 *** | 2.56 *** | 19.11 *** | 13070.89 *** |
| REPLICATION | 2 | 3668.67 ns | 1215.68 * | 0.07 ns | 1.54 ns | 2660.36 * |
| RESIDUALS | 200 | 1218.15 | 1383.90 | 0.16 | 2.83 | 3417.45 |
| H2 | 0.79 | 0.79 | 0.95 | 0.87 | 0.78 |
| Chr.Bin | Position | SNP-Alleles a | p-Value | Effect | Trait | BGSL |
|---|---|---|---|---|---|---|
| 1.02 | 18,282,139 | 2544389-10-G/C | 8.49 × 10−5 | −0.61926 | GWL | 0.3 |
| 1.02 | 21,511,322 | 2399751-6-C/A | 3.95 × 10−6 | 0.397572 | AP | 0.05 |
| 1.04 | 69,429,238 | 5584129-55-C/T | 1.20 × 10−13 | −1.34659 | AK | 0.01 |
| 1.04 | 69,747,754 | 4580363-8-A/G | 1.79 × 10−5 | −0.45677 | AK | 0.3 |
| 1.08 | 238,892,103 | 4583673-29-G/C | 6.29 × 10−6 | −0.45143 | GWL | 0.05 |
| 1.09 | 263,624,976 | 100024832-19-A/C | 3.83 × 10−7 | −1.39669 | GWL | 0.01 |
| 6.36 × 10−5 | −0.56467 | FAW | 0.3 | |||
| 1.09 | 264,933,475 | 4583685-9-G/A | 4.88 × 10−5 | −0.56303 | NH | 0.3 |
| 5.98 × 10−5 | −0.59983 | AK | 0.3 | |||
| 1.11 | 285,936,150 | 4580090-67-T/C | 8.37 × 10−7 | 0.796615 | GWL | 0.01 |
| 1.12 | 305,156,544 | 2382596-67-A/G | 1.63 × 10−10 | −0.36002 | FAW | 0.01 |
| 2.02 | 6,741,658 | 2452223-17-A/G | 6.79 × 10−6 | 0.213179 | FAW | 0.05 |
| 2.04 | 30,341,425 | 4771831-60-G/T | 2.22 × 10−6 | −0.61067 | AK | 0.01 |
| 2.04 | 35,377,279 | 4767220-53-G/A | 1.64 × 10−5 | −0.52503 | AK | 0.3 |
| 2.04 | 40,608,209 | 2388222-45-G/C | 1.01 × 10-9 | 0.473004 | FP | 0.01 |
| 1.35 × 10-9 | 0.752083 | GWL | 0.01 | |||
| 2.05 | 140,747,202 | 2435073-40-T/C | 3.13 × 10-6 | −0.41627 | AP | 0.01 |
| 7.09 × 10-5 | 0.289661 | FP | 0.3 | |||
| 2.06 | 154,630,564 | 2448649-48-G/A | 5.41 × 10-5 | −0.18627 | FAW | 0.3 |
| 2.08 | 213,714,960 | 4583437-30-G/C | 3.16 × 10-5 | 0.195599 | FP | 0.3 |
| 2.08 | 221,951,608 | 4765698-16-A/G | 2.42 × 10−5 | −1.02779 | AK | 0.3 |
| 2.10 | 236,778,497 | 100130818-44-A/G | 1.70 × 10−5 | 0.391072 | FP | 0.3 |
| 2.10 | 236,789,029 | 4591349-29-A/G | 5.73 × 10−6 | 0.366018 | GWL | 0.05 |
| 3.01 | 2,734,515 | 9714175-54-C/G | 1.74 × 10−8 | −0.52816 | FAW | 0.01 |
| 1.88 × 10−5 | 0.885829 | NH | 0.3 | |||
| 3.02 | 4,141,348 | 4764930-10-C/T | 3.43 × 10−7 | −0.51944 | FAW | 0.01 |
| 1.14 × 10−5 | −1.03199 | GWL | 0.05 | |||
| 3.04 | 17,591,392 | 4772102-17-T/G | 2.15 × 10−5 | 0.305217 | FP | 0.3 |
| 3.04 | 71,004,409 | 4593663-22-G/A | 2.15 × 10−10 | 0.966386 | GWL | 0.01 |
| 2.55 × 10−7 | 0.48656 | FP | 0.01 | |||
| 3.06 | 179,391,224 | 2446859-65-C/G | 9.98 × 10−6 | −0.30607 | AP | 0.05 |
| 3.07 | 201,766,146 | 4584446-12-G/C | 1.08 × 10−6 | 0.493102 | NH | 0.01 |
| 3.09 | 227,436,274 | 4583173-13-T/C | 9.21 × 10−6 | −0.62512 | GWL | 0.05 |
| 4.03 | 19,181,255 | 2381322-13-C/G | 5.34 × 10−9 | 0.202627 | FAW | 0.01 |
| 4.04 | 24,984,097 | 4779016-24-C/T | 5.56 × 10−5 | 0.468163 | NH | 0.3 |
| 4.05 | 48,323,977 | 4577027-47-G/A | 1.75 × 10−5 | 0.551228 | GWL | 0.3 |
| 4.05 | 78,882,987 | 100220678-45-A/G | 9.85 × 10−6 | 0.184883 | FAW | 0.05 |
| 7.12 × 10−5 | 0.324198 | FP | 0.3 | |||
| 4.08 | 180,072,262 | 4771330-29-T/C | 3.40 × 10−7 | −0.8024 | NH | 0.01 |
| 4.08 | 188,548,237 | 2619648-16-T/C | 3.79 × 10−6 | −0.91698 | GWL | 0.05 |
| 5.02 | 8,372,190 | 4589321-22-G/A | 7.07 × 10−6 | −0.67142 | AK | 0.05 |
| 5.03 | 32,460,125 | 7048960-37-T/G | 1.22 × 10−6 | −0.75462 | NH | 0.01 |
| 5.04 | 134,168,179 | 7049219-26-T/C | 5.10 × 10−5 | 0.16596 | FAW | 0.3 |
| 5.04 | 155,012,378 | 4584182-35-C/G | 2.64 × 10−5 | −0.16674 | FAW | 0.3 |
| 5.07 | 204,689,646 | 4774140-50-G/A | 1.51 × 10−5 | 0.372348 | FP | 0.05 |
| 6.01 | 9,188,598 | 4587005-7-C/G | 2.68 × 10−6 | −0.65839 | AK | 0.01 |
| 8.38 × 10−5 | −0.41379 | NH | 0.3 | |||
| 6.01 | 77,513,355 | 4771590-67-A/T | 4.69 × 10−5 | 0.299541 | FP | 0.3 |
| 6.03 | 103,106,812 | 5586936-13-T/C | 5.07 × 10−5 | 0.312373 | FP | 0.3 |
| 6.06 | 157,597,555 | 4579331-18-T/C | 1.90 × 10−6 | 0.544151 | AP | 0.01 |
| 6.08 | 169,246,523 | 4764931-6-G/A | 5.19 × 10−6 | −0.61458 | FP | 0.05 |
| 8.92 × 10−5 | −0.66184 | AP | 0.3 | |||
| 7.01 | 5,750,453 | 4771072-39-A/G | 6.80 × 10−5 | 0.428352 | GWL | 0.3 |
| 7.03 | 152,580,067 | 5587204-51-A/C | 2.45 × 10−5 | −1.30905 | AK | 0.3 |
| 7.05 | 173,989,867 | 4580355-27-G/A | 4.84 × 10−7 | −0.57199 | GWL | 0.01 |
| 5.08 × 10−7 | 0.406797 | AP | 0.01 | |||
| 8.00 | 328,928 | 4773640-63-T/A | 7.16 × 10−8 | 0.34059 | FP | 0.01 |
| 8.02 | 16,558,612 | 4770550-8-G/C | 6.47 × 10−6 | 0.374068 | GWL | 0.05 |
| 8.03 | 99,111,439 | 2504966-32-A/G | 9.62 × 10−6 | 0.264805 | FAW | 0.05 |
| 8.05 | 146,321,767 | 2559495-18-T/G | 6.26 × 10−5 | −0.15544 | FAW | 0.3 |
| 8.31 × 10−5 | −0.54446 | AK | 0.3 | |||
| 8.08 | 170,354,517 | 2610943-54-T/C | 3.08 × 10−6 | 0.570008 | GWL | 0.01 |
| 9.53 × 10−5 | −0.37079 | AP | 0.3 | |||
| 8.09 | 176,518,972 | 2376195-62-T/G | 7.58 × 10−5 | 0.393732 | FP | 0.3 |
| 8.09 | 180,177,242 | 4579847-66-T/G | 6.92 × 10−5 | 0.277417 | FP | 0.3 |
| 9.03 | 61,164,617 | 4771587-19-T/C | 6.24 × 10−7 | −1.9427 | AK | 0.01 |
| 9.04 | 120,457,334 | 100023814-29-T/G | 4.20 × 10−5 | −0.52317 | AK | 0.3 |
| 8.2 × 10−5 | 0.288404 | FP | 0.3 | |||
| 9.05 | 129,393,054 | 9682691-38-C/T | 5.68 × 10−10 | 0.797225 | FP | 0.01 |
| 4.41 × 10−6 | −0.34413 | FAW | 0.05 | |||
| 9.05 | 133,252,665 | 4764675-42-C/G | 2.69 × 10−5 | −0.52765 | AP | 0.3 |
| 9.06 | 141,396,844 | 2425091-21-G/A | 4.50 × 10−10 | 0.689575 | NH | 0.01 |
| 10.04 | 89,412,526 | 4582917-12-A/G | 1.05 × 10−7 | 0.571392 | GWL | 0.01 |
| 10.04 | 99,693,244 | 2539012-9-A/C | 2.39 × 10−10 | 0.767475 | GWL | 0.01 |
| 1.87 × 10−6 | 0.363841 | FP | 0.01 | |||
| 3.21 × 10−6 | −0.44639 | AP | 0.05 | |||
| 10.04 | 106,804,143 | 100298755-56-T/C | 2.20 × 10−8 | −0.31497 | FAW | 0.01 |
| 10.04 | 125,628,521 | 4776702-53-G/A | 1.89 × 10−7 | 1.750698 | AK | 0.01 |
| 10.05 | 136,798,456 | 7061499-37-A/G | 1.29 × 10−12 | 0.547738 | AP | 0.01 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badji, A.; Kwemoi, D.B.; Machida, L.; Okii, D.; Mwila, N.; Agbahoungba, S.; Kumi, F.; Ibanda, A.; Bararyenya, A.; Solemanegy, M.; et al. Genetic Basis of Maize Resistance to Multiple Insect Pests: Integrated Genome-Wide Comparative Mapping and Candidate Gene Prioritization. Genes 2020, 11, 689. https://doi.org/10.3390/genes11060689
Badji A, Kwemoi DB, Machida L, Okii D, Mwila N, Agbahoungba S, Kumi F, Ibanda A, Bararyenya A, Solemanegy M, et al. Genetic Basis of Maize Resistance to Multiple Insect Pests: Integrated Genome-Wide Comparative Mapping and Candidate Gene Prioritization. Genes. 2020; 11(6):689. https://doi.org/10.3390/genes11060689
Chicago/Turabian StyleBadji, A., D. B. Kwemoi, L. Machida, D. Okii, N. Mwila, S. Agbahoungba, F. Kumi, A. Ibanda, A. Bararyenya, M. Solemanegy, and et al. 2020. "Genetic Basis of Maize Resistance to Multiple Insect Pests: Integrated Genome-Wide Comparative Mapping and Candidate Gene Prioritization" Genes 11, no. 6: 689. https://doi.org/10.3390/genes11060689
APA StyleBadji, A., Kwemoi, D. B., Machida, L., Okii, D., Mwila, N., Agbahoungba, S., Kumi, F., Ibanda, A., Bararyenya, A., Solemanegy, M., Odong, T., Wasswa, P., Otim, M., Asea, G., Ochwo-Ssemakula, M., Talwana, H., Kyamanywa, S., & Rubaihayo, P. (2020). Genetic Basis of Maize Resistance to Multiple Insect Pests: Integrated Genome-Wide Comparative Mapping and Candidate Gene Prioritization. Genes, 11(6), 689. https://doi.org/10.3390/genes11060689