Analysis of the Hosts and Transmission Paths of SARS-CoV-2 in the COVID-19 Outbreak


Abstract
1. Introduction
2. Materials And Methods
2.1. Dataset
2.2. Natural Vector
2.3. Euclidean Distance and Hausdorff Distance
- The distance between two sets is always non-negative. The distance is zero if and only if the two sets are exactly the same.
- The direction doesn’t change the distance value, i.e., .
- The distance satisfies the triangular inequality, i.e., , for any X, Y, and Z.
2.4. Phylogenetic Study
2.5. Natural Graph
3. Results
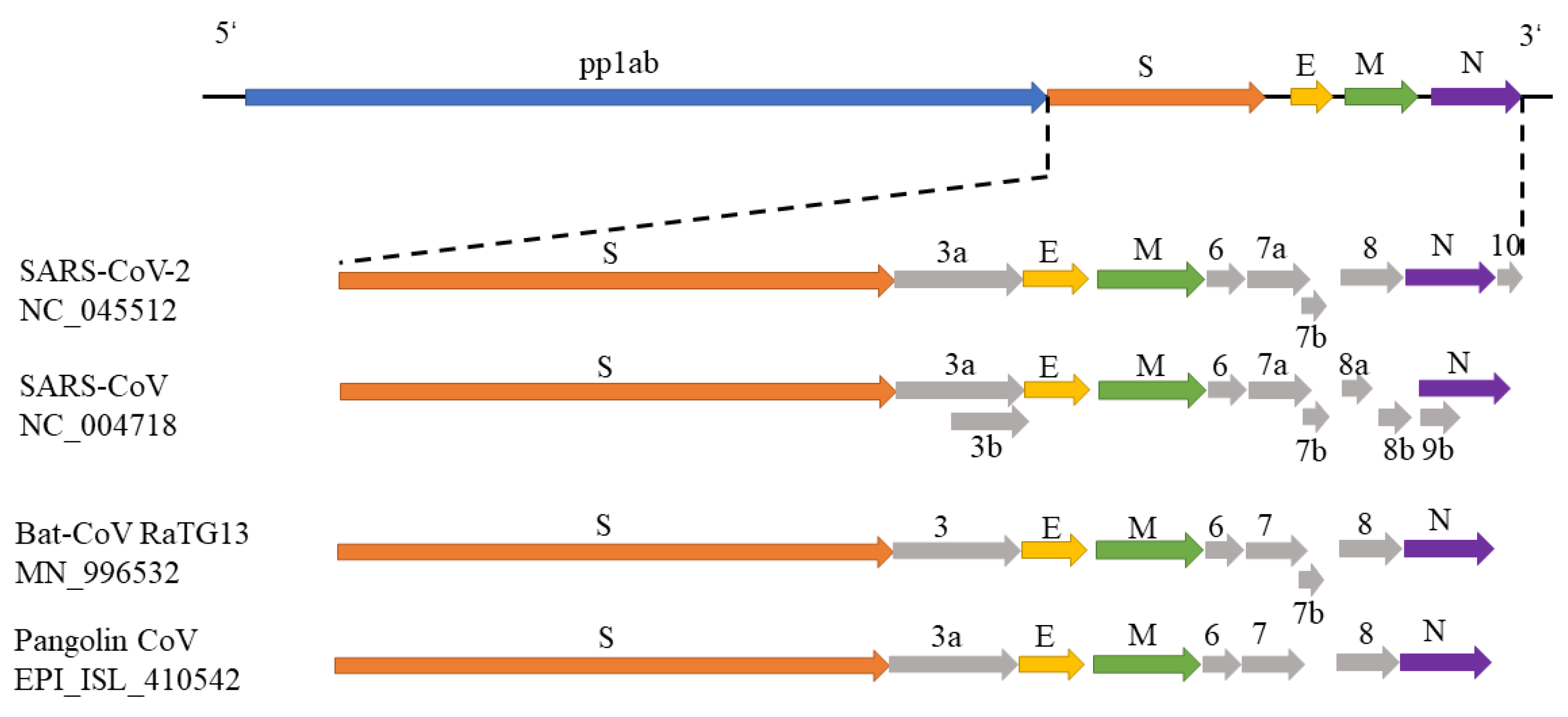
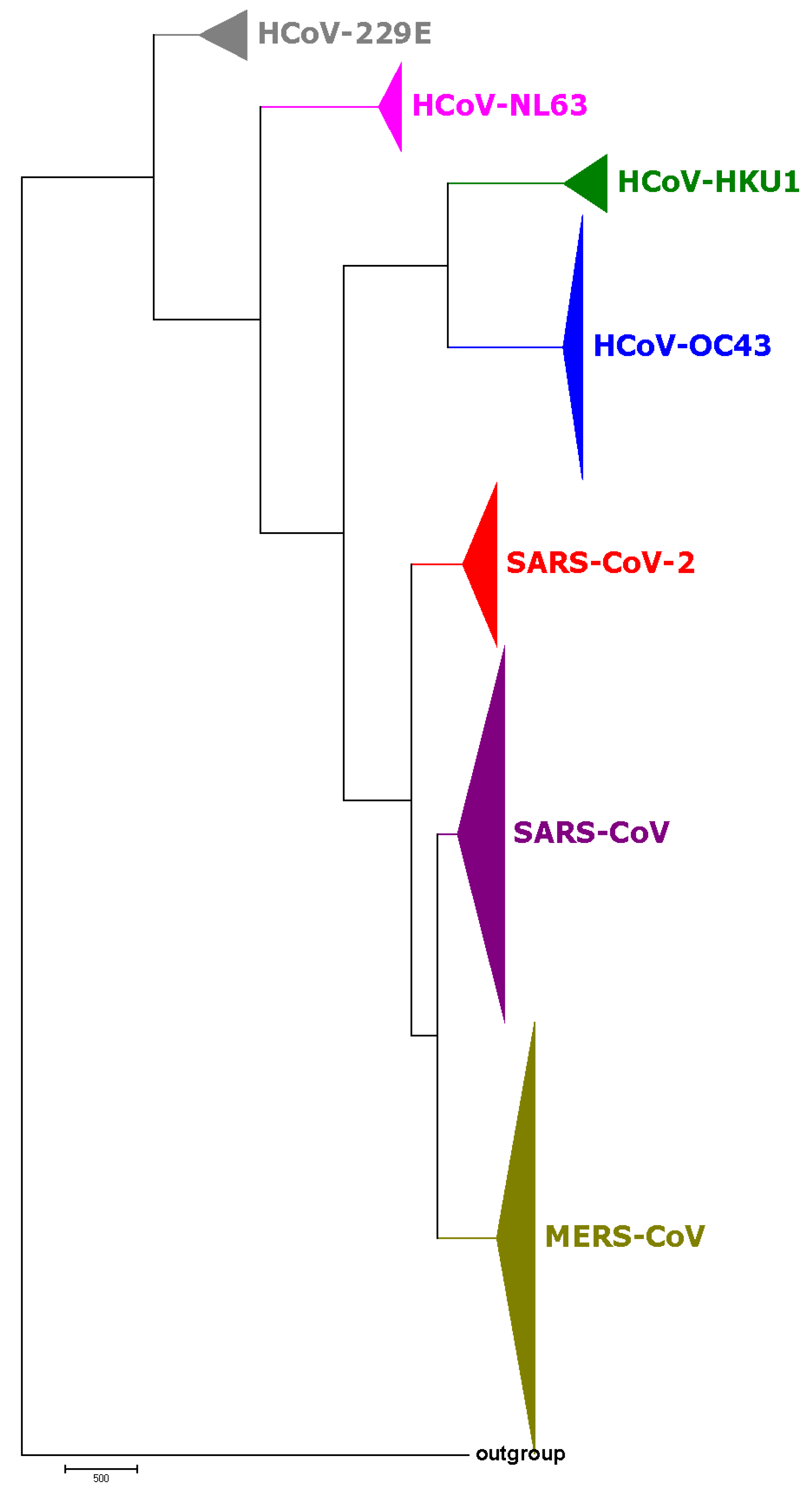
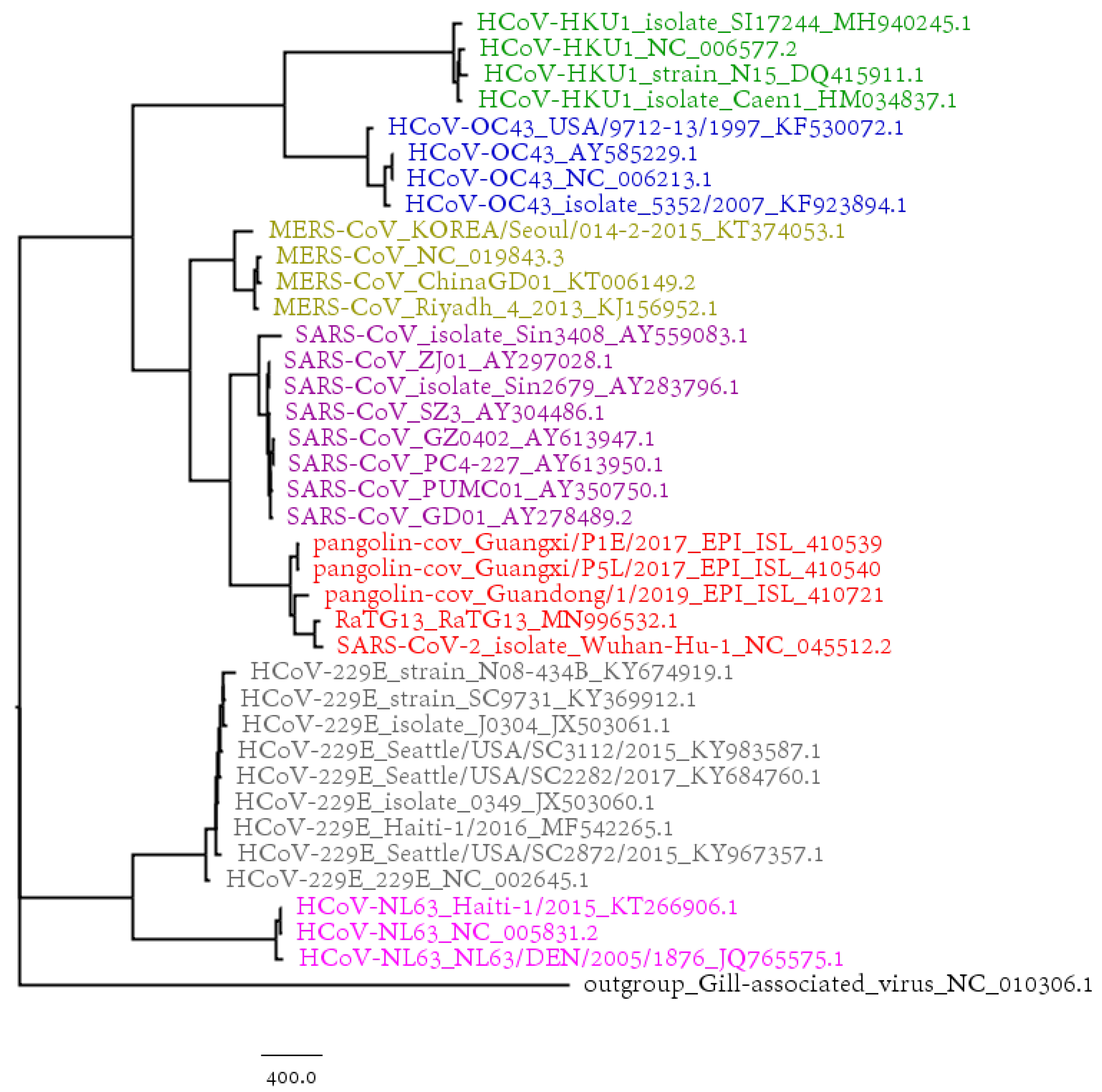
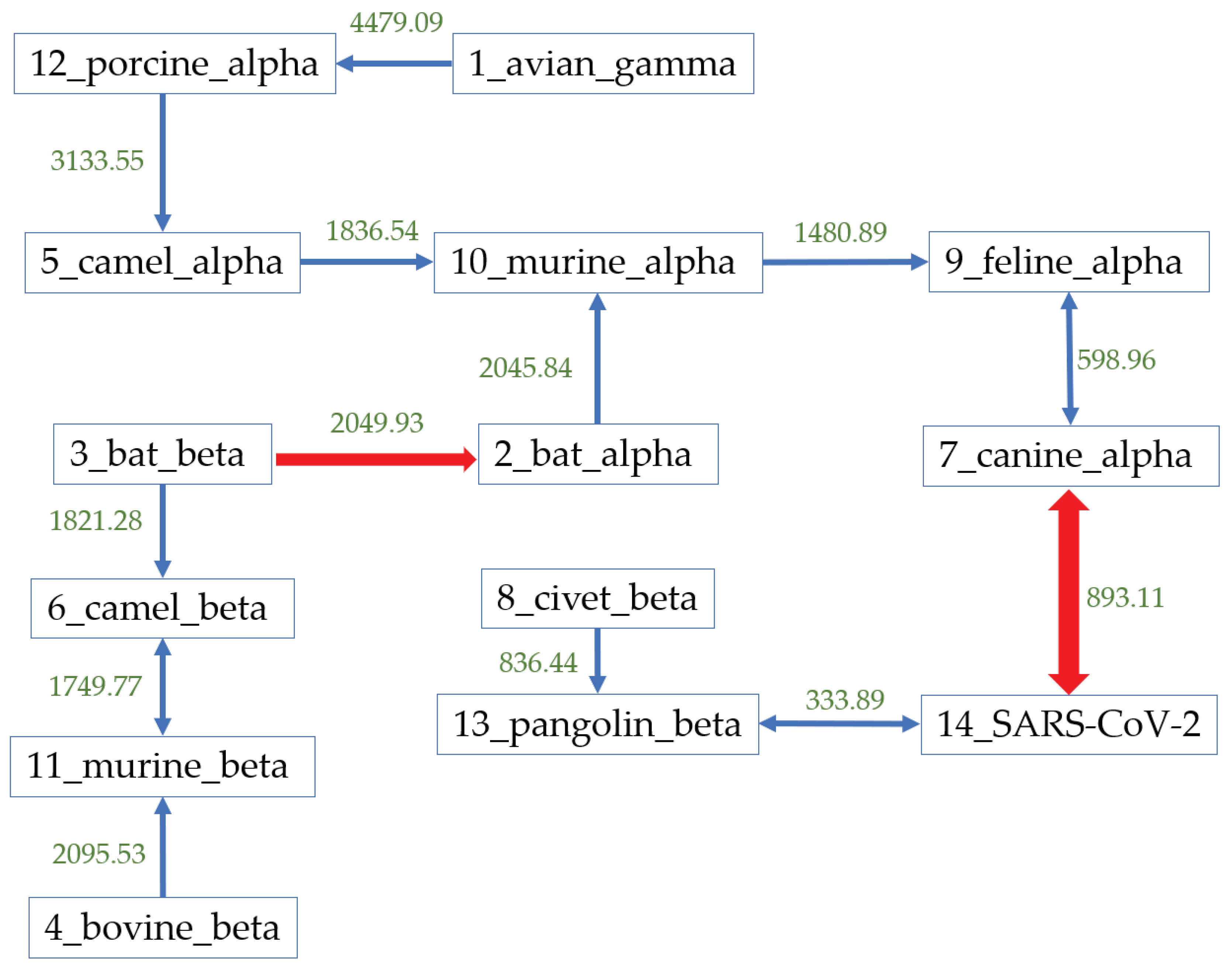
3.1. Phylogenetic Study of SARS-CoV-2
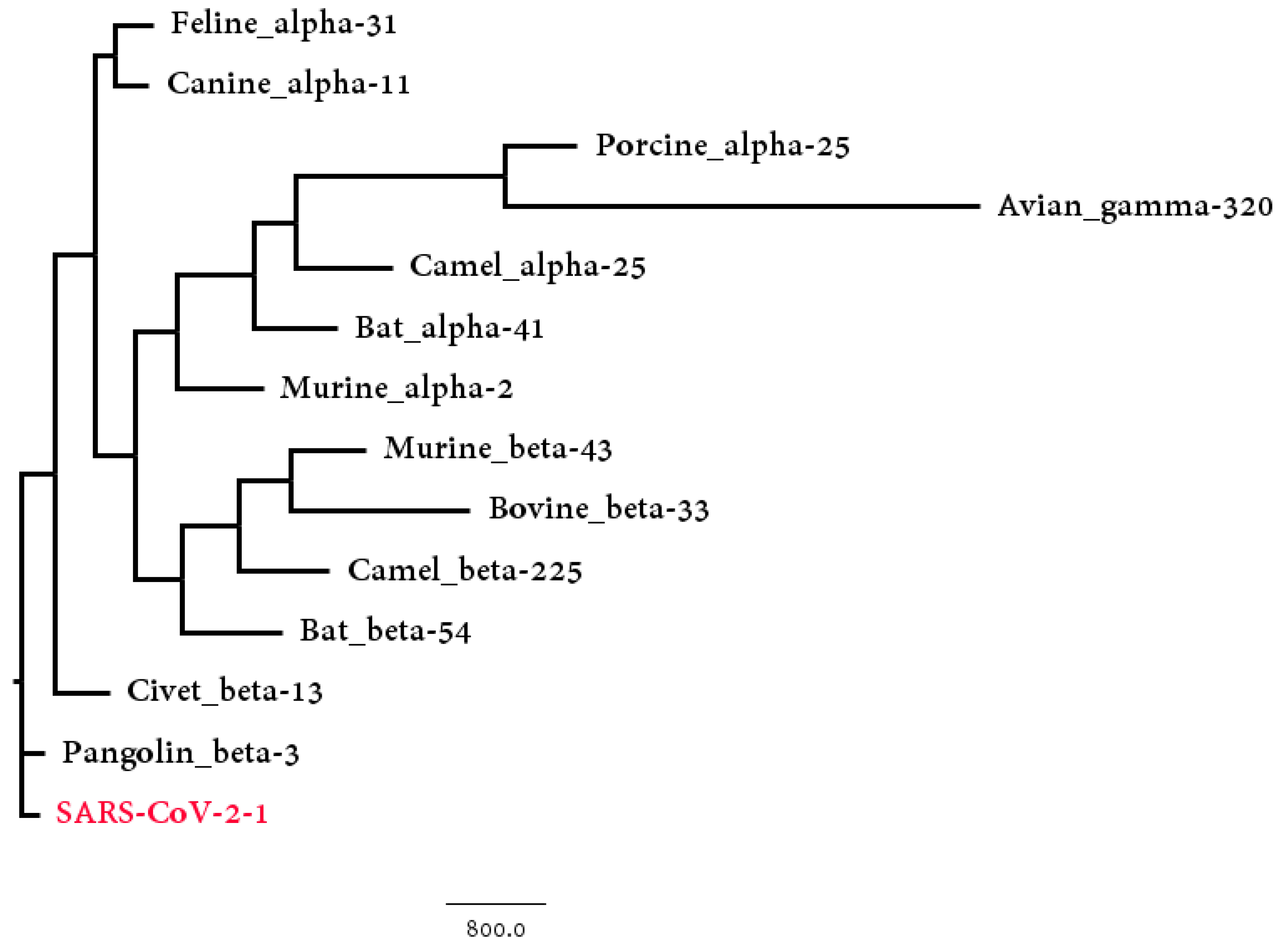
3.2. Host Identification of SARS-CoV-2
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Drosten, C.; Günther, S.; Preiser, W.; Van Der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. In Coronaviruses; Springer: Berlin/Heidelberg, Germany, 2015; pp. 1–23. [Google Scholar]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Jaimes, J.A.; André, N.M.; Chappie, J.S.; Millet, J.K.; Whittaker, G.R. Phylogenetic analysis and structural modeling of SARS-CoV-2 spike protein reveals an evolutionary distinct and proteolytically-sensitive activation loop. J. Mol. Biol. 2020, 432, 3309–3325. [Google Scholar] [CrossRef]
- Calligari, P.; Bobone, S.; Ricci, G.; Bocedi, A. Molecular investigation of SARS-CoV-2 proteins and their interactions with antiviral Drugs. Viruses 2020, 12, 445. [Google Scholar] [CrossRef]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.J.; Jiang, S. The spike protein of SARS-CoV—A target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar] [CrossRef]
- Lam, T.T.Y.; Shum, M.H.H.; Zhu, H.C.; Tong, Y.G.; Ni, X.B.; Liao, Y.S.; Wei, W.; Cheung, W.Y.M.; Li, W.J.; Li, L.F.; et al. Identifying SARS-CoV-2 related coronaviruses in Malayan pangolins. Nature 2020, 1–6. [Google Scholar] [CrossRef]
- Kan, B.; Wang, M.; Jing, H.; Xu, H.; Jiang, X.; Yan, M.; Liang, W.; Zheng, H.; Wan, K.; Liu, Q.; et al. Molecular evolution analysis and geographic investigation of severe acute respiratory syndrome coronavirus-like virus in palm civets at an animal market and on farms. J. Virol. 2005, 79, 11892–11900. [Google Scholar] [CrossRef]
- Ithete, N.L.; Stoffberg, S.; Corman, V.M.; Cottontail, V.M.; Richards, L.R.; Schoeman, M.C.; Drosten, C.; Drexler, J.F.; Preiser, W. Close relative of human Middle East respiratory syndrome coronavirus in bat, South Africa. Emerg. Infect. Dis. 2013, 19, 1697. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Y.; Li, J.L.; Yang, X.L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Zheng, B.; He, Y.; Liu, X.; Zhuang, Z.; Cheung, C.; Luo, S.; Li, P.; Zhang, L.; Guan, Y.; et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 2003, 302, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.; Woo, P.C.; Li, K.S.; Huang, Y.; Tsoi, H.W.; Wong, B.H.; Wong, S.S.; Leung, S.Y.; Chan, K.H.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. USA 2005, 102, 14040–14045. [Google Scholar] [CrossRef] [PubMed]
- Alagaili, A.N.; Briese, T.; Mishra, N.; Kapoor, V.; Sameroff, S.C.; de Wit, E.; Munster, V.J.; Hensley, L.E.; Zalmout, I.S.; Kapoor, A.; et al. Middle East respiratory syndrome coronavirus infection in dromedary camels in Saudi Arabia. MBio 2014, 5, e00884-14. [Google Scholar] [CrossRef] [PubMed]
- Forster, P.; Forster, L.; Renfrew, C.; Forster, M. Phylogenetic network analysis of SARS-CoV-2 genomes. Proc. Natl. Acad. Sci. USA 2020, 117, 9241–9243. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.R.; Cao, Q.D.; Hong, Z.S.; Tan, Y.Y.; Chen, S.D.; Jin, H.J.; Tan, K.S.; Wang, D.Y.; Yan, Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak–an update on the status. Mil. Med. Res. 2020, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Castillo, A.E.; Parra, B.; Tapia, P.; Acevedo, A.; Lagos, J.; Andrade, W.; Arata, L.; Leal, G.; Barra, G.; Tambley, C.; et al. Phylogenetic analysis of the first four SARS-CoV-2 cases in Chile. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Ji, W.; Wang, W.; Zhao, X.; Zai, J.; Li, X. Cross-species transmission of the newly identified coronavirus 2019-nCoV. J. Med. Virol. 2020, 92, 433–440. [Google Scholar] [CrossRef]
- Liu, Z.; Xiao, X.; Wei, X.; Li, J.; Yang, J.; Tan, H.; Zhu, J.; Zhang, Q.; Wu, J.; Liu, L. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS-CoV-2. J. Med. Virol. 2020, 92, 595–601. [Google Scholar] [CrossRef]
- Kiemer, L.; Lund, O.; Brunak, S.; Blom, N. Coronavirus 3CL pro proteinase cleavage sites: Possible relevance to SARS virus pathology. BMC Bioinform. 2004, 5, 72. [Google Scholar] [CrossRef]
- Jun, S.R.; Sims, G.E.; Wu, G.A.; Kim, S.H. Whole-proteome phylogeny of prokaryotes by feature frequency profiles: An alignment-free method with optimal feature resolution. Proc. Natl. Acad. Sci. USA 2010, 107, 133–138. [Google Scholar] [CrossRef]
- Yin, C.; Chen, Y.; Yau, S.S.T. A measure of DNA sequence similarity by Fourier transform with applications on hierarchical clustering. J. Theor. Biol. 2014, 359, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Zhu, Z.; Yin, C.; He, R.L.; Yau, S.S.T. A new method to cluster genomes based on cumulative Fourier power spectrum. Gene 2018, 673, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Pei, S.; Dong, R.; He, R.L.; Yau, S.S.T. Large-scale genome comparison based on cumulative Fourier power and phase spectra: Central moment and covariance vector. Comput. Struct. Biotechnol. J. 2019, 17, 982–994. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Yu, C.; Liang, Q.; He, R.L.; Yau, S.S.T. A novel method of characterizing genetic sequences: Genome space with biological distance and applications. PLoS ONE 2011, 6, e17293. [Google Scholar] [CrossRef]
- Huang, H.H.; Yu, C.; Zheng, H.; Hernandez, T.; Yau, S.C.; He, R.L.; Yang, J.; Yau, S.S.T. Global comparison of multiple-segmented viruses in 12-dimensional genome space. Mol. Phylogenet. Evol. 2014, 81, 29–36. [Google Scholar] [CrossRef]
- Zheng, H.; Yin, C.; Hoang, T.; He, R.L.; Yang, J.; Yau, S.S.T. Ebolavirus classification based on natural vectors. DNA Cell Biol. 2015, 34, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Zheng, H.; Tian, K.; Yau, S.C.; Mao, W.; Yu, W.; Yin, C.; Yu, C.; He, R.L.; Yang, J.; et al. Virus database and online inquiry system based on natural vectors. Evol. Bioinform. 2017, 13, 1176934317746667. [Google Scholar] [CrossRef]
- Yu, C.; He, R.L.; Yau, S.S.T. Viral genome phylogeny based on Lempel–Ziv complexity and Hausdorff distance. J. Theor. Biol. 2014, 348, 12–20. [Google Scholar] [CrossRef]
- Huttenlocher, D.P.; Klanderman, G.A.; Rucklidge, W.J. Comparing images using the Hausdorff distance. IEEE Trans. Pattern Anal. Mach. Intell. 1993, 15, 850–863. [Google Scholar] [CrossRef]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef]
- Desper, R.; Gascuel, O. Fast and accurate phylogeny reconstruction algorithms based on the minimum- evolution principle. In Proceedings of the International Workshop on Algorithms in Bioinformatics, Rome, Italy, 17–21 September 2002; Springer: Berlin/Heidelberg, Germany, 2002; pp. 357–374. [Google Scholar]
- Desper, R.; Gascuel, O. Theoretical foundation of the balanced minimum evolution method of phylogenetic inference and its relationship to weighted least-squares tree fitting. Mol. Biol. Evol. 2004, 21, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Gascuel, O. BIONJ: An improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 1997, 14, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef]
- Lacroix, A.; Duong, V.; Hul, V.; San, S.; Davun, H.; Omaliss, K.; Chea, S.; Hassanin, A.; Theppangna, W.; Silithammavong, S.; et al. Genetic diversity of coronaviruses in bats in Lao PDR and Cambodia. Infect. Genet. Evol. 2017, 48, 10–18. [Google Scholar] [CrossRef]
- Ren, W.; Li, W.; Yu, M.; Hao, P.; Zhang, Y.; Zhou, P.; Zhang, S.; Zhao, G.; Zhong, Y.; Wang, S.; et al. Full-length genome sequences of two SARS-like coronaviruses in horseshoe bats and genetic variation analysis. J. Gen. Virol. 2006, 87, 3355–3359. [Google Scholar] [CrossRef]
- Drexler, J.F.; Gloza-Rausch, F.; Glende, J.; Corman, V.M.; Muth, D.; Goettsche, M.; Seebens, A.; Niedrig, M.; Pfefferle, S.; Yordanov, S.; et al. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J. Virol. 2010, 84, 11336–11349. [Google Scholar] [CrossRef]
- Lau, S.K.; Li, K.S.; Huang, Y.; Shek, C.T.; Tse, H.; Wang, M.; Choi, G.K.; Xu, H.; Lam, C.S.; Guo, R.; et al. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J. Virol. 2010, 84, 2808–2819. [Google Scholar] [CrossRef]
- Rihtarič, D.; Hostnik, P.; Steyer, A.; Grom, J.; Toplak, I. Identification of SARS-like coronaviruses in horseshoe bats (Rhinolophus hipposideros) in Slovenia. Arch. Virol. 2010, 155, 507–514. [Google Scholar] [CrossRef]
- Yuan, J.; Hon, C.C.; Li, Y.; Wang, D.; Xu, G.; Zhang, H.; Zhou, P.; Poon, L.L.; Lam, T.T.Y.; Leung, F.C.C.; et al. Intraspecies diversity of SARS-like coronaviruses in Rhinolophus sinicus and its implications for the origin of SARS coronaviruses in humans. J. Gen. Virol. 2010, 91, 1058–1062. [Google Scholar] [CrossRef]
- Balboni, A.; Gallina, L.; Palladini, A.; Prosperi, S.; Battilani, M. A real-time PCR assay for bat SARS-like coronavirus detection and its application to Italian greater horseshoe bat faecal sample surveys. Sci. World J. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wu, Z.; Ren, X.; Yang, F.; He, G.; Zhang, J.; Dong, J.; Sun, L.; Zhu, Y.; Du, J.; et al. Novel SARS-like betacoronaviruses in bats, China, 2011. Emerg. Infect. Dis. 2013, 19, 989. [Google Scholar] [PubMed]
- He, B.; Zhang, Y.; Xu, L.; Yang, W.; Yang, F.; Feng, Y.; Xia, L.; Zhou, J.; Zhen, W.; Feng, Y.; et al. Identification of diverse alphacoronaviruses and genomic characterization of a novel severe acute respiratory syndrome-like coronavirus from bats in China. J. Virol. 2014, 88, 7070–7082. [Google Scholar] [CrossRef] [PubMed]
- Gouilh, M.A.; Puechmaille, S.J.; Gonzalez, J.P.; Teeling, E.; Kittayapong, P.; Manuguerra, J.C. SARS-Coronavirus ancestor’s foot-prints in South-East Asian bat colonies and the refuge theory. Infect. Genet. Evol. 2011, 11, 1690–1702. [Google Scholar] [CrossRef]
- Wacharapluesadee, S.; Duengkae, P.; Rodpan, A.; Kaewpom, T.; Maneeorn, P.; Kanchanasaka, B.; Yingsakmongkon, S.; Sittidetboripat, N.; Chareesaen, C.; Khlangsap, N.; et al. Diversity of coronavirus in bats from Eastern Thailand. Virol. J. 2015, 12, 57. [Google Scholar] [CrossRef]
- Tong, S.; Conrardy, C.; Ruone, S.; Kuzmin, I.V.; Guo, X.; Tao, Y.; Niezgoda, M.; Haynes, L.; Agwanda, B.; Breiman, R.F.; et al. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg. Infect. Dis. 2009, 15, 482. [Google Scholar] [CrossRef]
- Tian, K.; Zhao, X.; Zhang, Y.; Yau, S. Comparing protein structures and inferring functions with a novel three-dimensional Yau–Hausdorff method. J. Biomol. Struct. Dyn. 2019, 37, 4151–4160. [Google Scholar] [CrossRef]
- Phan, T. Genetic diversity and evolution of SARS-CoV-2. Infect. Genet. Evol. 2020, 81, 104260. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef]
- Zhang, T.; Wu, Q.; Zhang, Z. Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Current Biology 2020. [Google Scholar] [CrossRef]
- Zhang, C.; Zheng, W.; Huang, X.; Bell, E.W.; Zhou, X.; Zhang, Y. Protein structure and sequence re-analysis of 2019-nCoV genome refutes snakes as its intermediate host or the unique similarity between its spike protein insertions and HIV-1. J. Proteome Res. 2020, 19, 1351–1360. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3CL Proteinase | SARS-CoV-2 | SARS-CoV | HCoV-229E | MERS-CoV | HCoV-NL63 |
| PDB-Number | 6LU7 | 3AW0 | 2ZU2 | 5WKJ | 6FV2 |
| RMSD | 0.72 | 1.10 | 1.53 | 1.25 | |
| NV-Distance | 22.61 | 117.82 | 140.59 | 118.98 | |
| Spike Protein | SARS-CoV-2 | SARS-CoV | HCoV-229E | MERS-CoV | HCoV-NL63 |
| PDB-Number | 6VXX | 5X58 | 6U7H | 5X5F | 5SZS |
| RMSD | 1.74 | 2.21 | 3.20 | 2.71 | |
| NV-Distance | 235.39 | 349.86 | 289.54 | 401.99 |
| Host | Number | Hausdorff Distance | Center Distance | S-Protein Center Distance |
|---|---|---|---|---|
| Pangolin_beta | 3 | 333.89 | 230.11 | 117.39 |
| Civet_beta | 13 | 928.40 | 952.39 | 220.21 |
| Bat_beta | 54 | 2400.72 | 1102.03 | 205.47 |
| Murine_beta | 43 | 2620.43 | 2358.63 | 254.27 |
| Camel_beta | 225 | 2464.57 | 1307.54 | 353.01 |
| Bovine_beta | 33 | 2571.48 | 2377.34 | 317.78 |
| Avian_gamma | 320 | 8788.62 | 2753.43 | 426.91 |
| Bat_alpha | 41 | 3340.69 | 2494.54 | 257.71 |
| Camel_alpha | 25 | 3065.11 | 3044.61 | 405.31 |
| Canine_alpha | 11 | 893.11 | 947.82 | 485.56 |
| Feline_alpha | 31 | 1205.55 | 107.93 | 453.74 |
| Murine_alpha | 2 | 2168.66 | 2125.31 | 482.57 |
| Porcine_alpha | 25 | 4981.11 | 2784.12 | 391.29 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, R.; Pei, S.; Yin, C.; He, R.L.; Yau, S.S.-T. Analysis of the Hosts and Transmission Paths of SARS-CoV-2 in the COVID-19 Outbreak. Genes 2020, 11, 637. https://doi.org/10.3390/genes11060637
Dong R, Pei S, Yin C, He RL, Yau SS-T. Analysis of the Hosts and Transmission Paths of SARS-CoV-2 in the COVID-19 Outbreak. Genes. 2020; 11(6):637. https://doi.org/10.3390/genes11060637
Chicago/Turabian StyleDong, Rui, Shaojun Pei, Changchuan Yin, Rong Lucy He, and Stephen S.-T. Yau. 2020. "Analysis of the Hosts and Transmission Paths of SARS-CoV-2 in the COVID-19 Outbreak" Genes 11, no. 6: 637. https://doi.org/10.3390/genes11060637
APA StyleDong, R., Pei, S., Yin, C., He, R. L., & Yau, S. S.-T. (2020). Analysis of the Hosts and Transmission Paths of SARS-CoV-2 in the COVID-19 Outbreak. Genes, 11(6), 637. https://doi.org/10.3390/genes11060637