Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability

 , ,
, , 
Abstract
1. Introduction
2. HDACs in Homo sapiens and Other Organisms
3. HDAC Specificity
3.1. Enzymatic Specificity of HDAC Activity
3.1.1. Class I: (HDAC1, -2, -3, and -8)
3.1.2. Class IIa: (HDAC4, -5, -7, and -9)
3.1.3. Class IIb: (HDACs -6 and -10)
3.1.4. Class IV: (HDAC11)
3.2. Tissue Specificity of HDACs
4. HDACs in Transcriptional Complexes
4.1. Sin3 Complex
4.2. Co Rest Complex
4.3. NuRD Complex
4.4. MiDac Complex
4.5. SMRT Complex
4.6. Class IIa HDACs
4.7. Class IV HDACs
5. Pan-Cancer Analysis of HDACs
6. HDAC Inhibitors as Anticancer Drugs
6.1. HDAC Inhibitors
6.2. Group I—Hydroxamic Acids
6.2.1. PAN-Hydroxamic Acid Compounds
6.2.2. Specific Hydroxamic Acid Compounds
6.3. Group II—Short-Chain Fatty (Aliphatic) Acids
6.4. Group III—Benzamides
6.5. Group IV—Cyclic Peptides
7. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CoR | Nuclear Receptor Co-Repressor |
| COVID-19 | CoronaVirus Disease 2019 |
| DAD | Deacetylase-Activating Domain |
| EMT | Epithelial-to-Mesenchymal Transition |
| FDA | Food and Drug Administration |
| FK228 | Romidepsin |
| FPKM | Fragments Per Kilobase of transcript length per Million of mapped reads |
| GTEx | Genotype-Tissue Expression project |
| HAT | Histone Acetyltransferase |
| HCC | Hepatocellular Carcinoma |
| HDAC | Histone Deacetylase |
| HDACi | Histone Deacetylase inhibitor |
| IFN | Interferon |
| LBH589 | Panobinostat |
| LSD1 | Lysine Demethylase 1 |
| MEF2 | Myocyte Enhancer Factor 2 |
| NES | Nuclear Exporting Signal |
| MGCD0103 | Mocetinostat |
| MiDac | Mitotic Deacetylase Complex |
| MS-275 | Entinostat |
| NBL | Neuroblastoma |
| NLS | Nuclear Localization Sequence |
| NR | Nuclear Receptor |
| NuRD | Nucleosome Remodeling Deacetylase |
| PTM | Post-Translational Modification |
| PXD101 | Belinostat |
| RD | Repression Domain |
| REST | RE1 Silencing Transcription Factor |
| SAHA | Vorinostat |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome CoronaVirus 2 |
| SIRT | Sirtuins |
| SMN | Survival of MotoNeurons |
| TCGA | The Cancer Genome Atlas |
| TF | Transcription Factor |
| TNBS | Triple Negative Breast Cancer |
| TSA | Thrichostatin A |
| TSS | Transcription Start Site |
| VPA | Valproic Acid |
References
- Gregoretti, I.V.; Lee, Y.-M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Ceccacci, E.; Minucci, S. Inhibition of histone deacetylases in cancer therapy: Lessons from leukaemia. Br. J. Cancer 2016, 114, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Mercatelli, D.; Scalambra, L.; Triboli, L.; Ray, F.; Giorgi, F.M. Gene regulatory network inference resources: A practical overview. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194430. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef]
- Karpac, J.; Jasper, H. Metabolic homeostasis: HDACs take center stage. Cell 2011, 145, 497–499. [Google Scholar] [CrossRef]
- Chuang, D.-M.; Leng, Y.; Marinova, Z.; Kim, H.-J.; Chiu, C.-T. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009, 32, 591–601. [Google Scholar] [CrossRef]
- Consalvi, S.; Saccone, V.; Giordani, L.; Minetti, G.; Mozzetta, C.; Puri, P.L. Histone deacetylase inhibitors in the treatment of muscular dystrophies: Epigenetic drugs for genetic diseases. Mol. Med. 2011, 17, 457–465. [Google Scholar] [CrossRef]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef]
- Bheda, P.; Jing, H.; Wolberger, C.; Lin, H. The Substrate Specificity of Sirtuins. Annu. Rev. Biochem. 2016, 85, 405–429. [Google Scholar] [CrossRef]
- Yang, X.-J.; Seto, E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef]
- Fujimoto, D. Specificities of histone deacetylases from several animal and plant tissues. J. Biochem. 1972, 72, 1269–1271. [Google Scholar] [CrossRef]
- Boffa, L.C.; Vidali, G.; Mann, R.S.; Allfrey, V.G. Suppression of histone deacetylation in vivo and in vitro by sodium butyrate. J. Biol. Chem. 1978, 253, 3364–3366. [Google Scholar]
- Alonso, W.R.; Nelson, D.A. A novel yeast histone deacetylase: Partial characterization and development of an activity assay. Biochim. Biophys. Acta 1986, 866, 161–169. [Google Scholar] [CrossRef]
- Rundlett, S.E.; Carmen, A.A.; Kobayashi, R.; Bavykin, S.; Turner, B.M.; Grunstein, M. HDA1 and RPD3 are members of distinct yeast histone deacetylase complexes that regulate silencing and transcription. Proc. Natl. Acad. Sci. USA 1996, 93, 14503–14508. [Google Scholar] [CrossRef] [PubMed]
- Taunton, J.; Hassig, C.A.; Schreiber, S.L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 1996, 272, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Hassig, C.A.; Schreiber, S.L. Nuclear histone acetylases and deacetylases and transcriptional regulation: HATs off to HDACs. Curr. Opin. Chem. Biol. 1997, 1, 300–308. [Google Scholar] [CrossRef]
- Bartl, S.; Taplick, J.; Lagger, G.; Khier, H.; Kuchler, K.; Seiser, C. Identification of mouse histone deacetylase 1 as a growth factor-inducible gene. Mol. Cell. Biol. 1997, 17, 5033–5043. [Google Scholar] [CrossRef]
- Stiegler, P.; De Luca, A.; Bagella, L.; Giordano, A. The COOH-terminal region of pRb2/p130 binds to histone deacetylase 1 (HDAC1), enhancing transcriptional repression of the E2F-dependent cyclin A promoter. Cancer Res. 1998, 58, 5049–5052. [Google Scholar]
- Betz, R.; Gray, S.G.; Ekström, C.; Larsson, C.; Ekström, T.J. Human histone deacetylase 2, HDAC2 (Human RPD3), is localized to 6q21 by radiation hybrid mapping. Genomics 1998, 52, 245–246. [Google Scholar] [CrossRef]
- Emiliani, S.; Fischle, W.; Van Lint, C.; Al-Abed, Y.; Verdin, E. Characterization of a human RPD3 ortholog, HDAC3. Proc. Natl. Acad. Sci. USA 1998, 95, 2795–2800. [Google Scholar] [CrossRef]
- Grozinger, C.M.; Hassig, C.A.; Schreiber, S.L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. USA 1999, 96, 4868–4873. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.Y.; Ji, Y.J.; Jee, C.; Kim, D.H.; Ahnn, J. Characterization of CeHDA-7, a class II histone deacetylase interacting with MEF-2 in Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 2002, 293, 1295–1300. [Google Scholar] [CrossRef]
- Johnson, C.A.; Barlow, A.L.; Turner, B.M. Molecular cloning of Drosophila melanogaster cDNAs that encode a novel histone deacetylase dHDAC3. Gene 1998, 221, 127–134. [Google Scholar] [CrossRef]
- He, Y.; Tang, D.; Li, W.; Chai, R.; Li, H. Histone deacetylase 1 is required for the development of the zebrafish inner ear. Sci. Rep. 2016, 6, 16535. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Chen, W.; Qin, Y.; Huang, L.; Xu, X.; Zhao, L.; Yan, Q. AcuC, a histone deacetylase, contributes to the pathogenicity of Aeromonas hydrophila. Microbiologyopen 2017, 6, e00468. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Tong, J.K.; Schreiber, S.L. Genomewide studies of histone deacetylase function in yeast. Proc. Natl. Acad. Sci. USA 2000, 97, 13708–13713. [Google Scholar] [CrossRef]
- Bürger, M.; Chory, J. Structural and chemical biology of deacetylases for carbohydrates, proteins, small molecules and histones. Commun. Biol. 2018, 1, 217. [Google Scholar] [CrossRef]
- Guarente, L. Sir2 links chromatin silencing, metabolism, and aging. Genes Dev. 2000, 14, 1021–1026. [Google Scholar]
- Lundberg, K.S.; Shoemaker, D.D.; Adams, M.W.; Short, J.M.; Sorge, J.A.; Mathur, E.J. High-fidelity amplification using a thermostable DNA polymerase isolated from Pyrococcus furiosus. Gene 1991, 108, 1–6. [Google Scholar] [CrossRef]
- Henneman, B.; van Emmerik, C.; van Ingen, H.; Dame, R.T. Structure and function of archaeal histones. PLoS Genet. 2018, 14, e1007582. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Zuckerkandl, E.; Pauling, L. Evolutionary Divergence and Convergence in Proteins. In Evolving Genes and Proteins; Bryson, V., Vogel, H.J., Eds.; Academic Press: Cambridge, MA, USA, 1965; pp. 97–166. ISBN 978-1-4832-2734-4. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Amemiya, C.T.; Alföldi, J.; Lee, A.P.; Fan, S.; Philippe, H.; Maccallum, I.; Braasch, I.; Manousaki, T.; Schneider, I.; Rohner, N.; et al. The African coelacanth genome provides insights into tetrapod evolution. Nature 2013, 496, 311–316. [Google Scholar] [CrossRef]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef]
- Zou, H.; Wu, Y.; Navre, M.; Sang, B.-C. Characterization of the two catalytic domains in histone deacetylase 6. Biochem. Biophys. Res. Commun. 2006, 341, 45–50. [Google Scholar] [CrossRef]
- Backs, J.; Worst, B.C.; Lehmann, L.H.; Patrick, D.M.; Jebessa, Z.; Kreusser, M.M.; Sun, Q.; Chen, L.; Heft, C.; Katus, H.A.; et al. Selective repression of MEF2 activity by PKA-dependent proteolysis of HDAC4. J. Cell Biol. 2011, 195, 403–415. [Google Scholar] [CrossRef]
- Ginnan, R.; Sun, L.Y.; Schwarz, J.J.; Singer, H.A. MEF2 is regulated by CaMKIIδ2 and a HDAC4-HDAC5 heterodimer in vascular smooth muscle cells. Biochem. J. 2012, 444, 105–114. [Google Scholar] [CrossRef]
- McKinsey, T.A.; Zhang, C.L.; Olson, E.N. Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc. Natl. Acad. Sci. USA 2000, 97, 14400–14405. [Google Scholar] [CrossRef]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.-F.; Yao, T.-P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Taplick, J.; Kurtev, V.; Kroboth, K.; Posch, M.; Lechner, T.; Seiser, C. Homo-oligomerisation and nuclear localisation of mouse histone deacetylase 1. J. Mol. Biol. 2001, 308, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-M.; Tsai, S.-C.; Wen, Y.-D.; Fejer, G.; Seto, E. Functional domains of histone deacetylase-3. J. Biol. Chem. 2002, 277, 9447–9454. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Derfoul, A.; Pereira-Mouries, L.; Hall, D.J. A novel domain in histone deacetylase 1 and 2 mediates repression of cartilage-specific genes in human chondrocytes. FASEB J. 2009, 23, 3539–3552. [Google Scholar] [CrossRef]
- Park, H.; Kim, Y.; Park, D.; Jeoung, D. Nuclear localization signal domain of HDAC3 is necessary and sufficient for the expression regulation of MDR1. BMB Rep. 2014, 47, 342–347. [Google Scholar] [CrossRef]
- Lehmann, M.; Hoffmann, M.J.; Koch, A.; Ulrich, S.M.; Schulz, W.A.; Niegisch, G. Histone deacetylase 8 is deregulated in urothelial cancer but not a target for efficient treatment. J. Exp. Clin. Cancer Res. 2014, 33, 59. [Google Scholar] [CrossRef]
- Wang, A.H.; Yang, X.J. Histone deacetylase 4 possesses intrinsic nuclear import and export signals. Mol. Cell. Biol. 2001, 21, 5992–6005. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- McGinty, R.K.; Tan, S. Nucleosome structure and function. Chem. Rev. 2015, 115, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Margueron, R.; Hu, G.; Stokes, D.; Wang, Y.-H.; Reinberg, D. Highly compacted chromatin formed in vitro reflects the dynamics of transcription activation in vivo. Mol. Cell 2010, 38, 41–53. [Google Scholar] [CrossRef]
- Eberharter, A.; Becker, P.B. Histone acetylation: A switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. 2002, 3, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-J.; Seto, E. HATs and HDACs: From structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007, 26, 5310–5318. [Google Scholar] [CrossRef] [PubMed]
- de Ruijter, A.J.M.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B.P. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Ganai, S.A. Histone Deacetylase Inhibitors Modulating Non-epigenetic Players: The Novel Mechanism for Small Molecule Based Therapeutic Intervention. Curr. Drug Targets 2018, 19, 593–601. [Google Scholar] [CrossRef]
- Roos, W.P.; Krumm, A. The multifaceted influence of histone deacetylases on DNA damage signalling and DNA repair. Nucleic Acids Res. 2016, 44, 10017–10030. [Google Scholar] [CrossRef]
- Lahue, R.S.; Frizzell, A. Histone deacetylase complexes as caretakers of genome stability. Epigenetics 2012, 7, 806–810. [Google Scholar] [CrossRef]
- Robert, T.; Vanoli, F.; Chiolo, I.; Shubassi, G.; Bernstein, K.A.; Rothstein, R.; Botrugno, O.A.; Parazzoli, D.; Oldani, A.; Minucci, S.; et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011, 471, 74–79. [Google Scholar] [CrossRef]
- Guzzi, P.H.; Mercatelli, D.; Ceraolo, C.; Giorgi, F.M. Master Regulator Analysis of the SARS-CoV-2/Human Interactome. JCM 2020, 9, 982. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020. [Google Scholar] [CrossRef]
- Stobart, C.C.; Sexton, N.R.; Munjal, H.; Lu, X.; Molland, K.L.; Tomar, S.; Mesecar, A.D.; Denison, M.R. Chimeric Exchange of Coronavirus nsp5 Proteases (3CLpro) Identifies Common and Divergent Regulatory Determinants of Protease Activity. J. Virol. 2013, 87, 12611–12618. [Google Scholar] [CrossRef]
- Somoza, J.R.; Skene, R.J.; Katz, B.A.; Mol, C.; Ho, J.D.; Jennings, A.J.; Luong, C.; Arvai, A.; Buggy, J.J.; Chi, E.; et al. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 2004, 12, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Olson, D.E.; Udeshi, N.D.; Wolfson, N.A.; Pitcairn, C.A.; Sullivan, E.D.; Jaffe, J.D.; Svinkina, T.; Natoli, T.; Lu, X.; Paulk, J.; et al. An unbiased approach to identify endogenous substrates of “histone” deacetylase 8. ACS Chem. Biol. 2014, 9, 2210–2216. [Google Scholar] [CrossRef]
- Lopez, J.E.; Haynes, S.E.; Majmudar, J.D.; Martin, B.R.; Fierke, C.A. HDAC8 Substrates Identified by Genetically Encoded Active Site Photocrosslinking. J. Am. Chem. Soc. 2017, 139, 16222–16227. [Google Scholar] [CrossRef]
- Li, T.; Song, B.; Wu, Z.; Lu, M.; Zhu, W.-G. Systematic identification of Class I HDAC substrates. Brief. Bioinform. 2014, 15, 963–972. [Google Scholar] [CrossRef]
- Itoh, T.; Fairall, L.; Muskett, F.W.; Milano, C.P.; Watson, P.J.; Arnaudo, N.; Saleh, A.; Millard, C.J.; El-Mezgueldi, M.; Martino, F.; et al. Structural and functional characterization of a cell cycle associated HDAC1/2 complex reveals the structural basis for complex assembly and nucleosome targeting. Nucleic Acids Res. 2015, 43, 2033–2044. [Google Scholar] [CrossRef]
- Millard, C.J.; Watson, P.J.; Celardo, I.; Gordiyenko, Y.; Cowley, S.M.; Robinson, C.V.; Fairall, L.; Schwabe, J.W.R. Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol. Cell 2013, 51, 57–67. [Google Scholar] [CrossRef]
- Watson, P.J.; Millard, C.J.; Riley, A.M.; Robertson, N.S.; Wright, L.C.; Godage, H.Y.; Cowley, S.M.; Jamieson, A.G.; Potter, B.V.L.; Schwabe, J.W.R. Insights into the activation mechanism of class I HDAC complexes by inositol phosphates. Nat. Commun. 2016, 7, 11262. [Google Scholar] [CrossRef]
- Ito, A.; Kawaguchi, Y.; Lai, C.-H.; Kovacs, J.J.; Higashimoto, Y.; Appella, E.; Yao, T.-P. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002, 21, 6236–6245. [Google Scholar] [CrossRef]
- Martínez-Balbás, M.A.; Bauer, U.M.; Nielsen, S.J.; Brehm, A.; Kouzarides, T. Regulation of E2F1 activity by acetylation. EMBO J. 2000, 19, 662–671. [Google Scholar] [CrossRef]
- Yao, Y.L.; Yang, W.M.; Seto, E. Regulation of transcription factor YY1 by acetylation and deacetylation. Mol. Cell. Biol. 2001, 21, 5979–5991. [Google Scholar] [CrossRef] [PubMed]
- Naryzhny, S.N.; Lee, H. The post-translational modifications of proliferating cell nuclear antigen: Acetylation, not phosphorylation, plays an important role in the regulation of its function. J. Biol. Chem. 2004, 279, 20194–20199. [Google Scholar] [CrossRef] [PubMed]
- Nalawansha, D.A.; Pflum, M.K.H. LSD1 Substrate Binding and Gene Expression Are Affected by HDAC1-Mediated Deacetylation. ACS Chem. Biol. 2017, 12, 254–264. [Google Scholar] [CrossRef]
- Nalawansha, D.A.; Zhang, Y.; Herath, K.; Pflum, M.K.H. HDAC1 Substrate Profiling Using Proteomics-Based Substrate Trapping. ACS Chem. Biol. 2018, 13, 3315–3324. [Google Scholar] [CrossRef]
- Wilson, B.J.; Tremblay, A.M.; Deblois, G.; Sylvain-Drolet, G.; Giguère, V. An acetylation switch modulates the transcriptional activity of estrogen-related receptor alpha. Mol. Endocrinol. 2010, 24, 1349–1358. [Google Scholar] [CrossRef]
- Deardorff, M.A.; Bando, M.; Nakato, R.; Watrin, E.; Itoh, T.; Minamino, M.; Saitoh, K.; Komata, M.; Katou, Y.; Clark, D.; et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 2012, 489, 313–317. [Google Scholar] [CrossRef]
- Aramsangtienchai, P.; Spiegelman, N.A.; He, B.; Miller, S.P.; Dai, L.; Zhao, Y.; Lin, H. HDAC8 Catalyzes the Hydrolysis of Long Chain Fatty Acyl Lysine. ACS Chem. Biol. 2016, 11, 2685–2692. [Google Scholar] [CrossRef]
- Wei, W.; Liu, X.; Chen, J.; Gao, S.; Lu, L.; Zhang, H.; Ding, G.; Wang, Z.; Chen, Z.; Shi, T.; et al. Class I histone deacetylases are major histone decrotonylases: Evidence for critical and broad function of histone crotonylation in transcription. Cell Res. 2017, 27, 898–915. [Google Scholar] [CrossRef]
- Chawla, S.; Vanhoutte, P.; Arnold, F.J.L.; Huang, C.L.-H.; Bading, H. Neuronal activity-dependent nucleocytoplasmic shuttling of HDAC4 and HDAC5. J. Neurochem. 2003, 85, 151–159. [Google Scholar] [CrossRef]
- Di Giorgio, E.; Brancolini, C. Regulation of class IIa HDAC activities: It is not only matter of subcellular localization. Epigenomics 2016, 8, 251–269. [Google Scholar] [CrossRef]
- Dressel, U.; Bailey, P.J.; Wang, S.C.; Downes, M.; Evans, R.M.; Muscat, G.E. A dynamic role for HDAC7 in MEF2-mediated muscle differentiation. J. Biol. Chem. 2001, 276, 17007–17013. [Google Scholar] [CrossRef]
- McKinsey, T.A.; Zhang, C.L.; Lu, J.; Olson, E.N. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 2000, 408, 106–111. [Google Scholar] [CrossRef]
- Lu, J.; McKinsey, T.A.; Zhang, C.L.; Olson, E.N. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol. Cell 2000, 6, 233–244. [Google Scholar] [CrossRef]
- Lahm, A.; Paolini, C.; Pallaoro, M.; Nardi, M.C.; Jones, P.; Neddermann, P.; Sambucini, S.; Bottomley, M.J.; Lo Surdo, P.; Carfí, A.; et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. USA 2007, 104, 17335–17340. [Google Scholar] [CrossRef]
- Zhang, C.L.; McKinsey, T.A.; Olson, E.N. The transcriptional corepressor MITR is a signal-responsive inhibitor of myogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 7354–7359. [Google Scholar] [CrossRef]
- Zhou, X.; Marks, P.A.; Rifkind, R.A.; Richon, V.M. Cloning and characterization of a histone deacetylase, HDAC9. Proc. Natl. Acad. Sci. USA 2001, 98, 10572–10577. [Google Scholar] [CrossRef]
- Lobera, M.; Madauss, K.P.; Pohlhaus, D.T.; Wright, Q.G.; Trocha, M.; Schmidt, D.R.; Baloglu, E.; Trump, R.P.; Head, M.S.; Hofmann, G.A.; et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol. 2013, 9, 319–325. [Google Scholar] [CrossRef]
- Jeon, E.-J.; Lee, K.-Y.; Choi, N.-S.; Lee, M.-H.; Kim, H.-N.; Jin, Y.-H.; Ryoo, H.-M.; Choi, J.-Y.; Yoshida, M.; Nishino, N.; et al. Bone morphogenetic protein-2 stimulates Runx2 acetylation. J. Biol. Chem. 2006, 281, 16502–16511. [Google Scholar] [CrossRef]
- Luo, L.; Martin, S.C.; Parkington, J.; Cadena, S.M.; Zhu, J.; Ibebunjo, C.; Summermatter, S.; Londraville, N.; Patora-Komisarska, K.; Widler, L.; et al. HDAC4 Controls Muscle Homeostasis through Deacetylation of Myosin Heavy Chain, PGC-1α, and Hsc70. Cell Rep. 2019, 29, 749–763. [Google Scholar] [CrossRef]
- Verdel, A.; Curtet, S.; Brocard, M.P.; Rousseaux, S.; Lemercier, C.; Yoshida, M.; Khochbin, S. Active maintenance of mHDA2/mHDAC6 histone-deacetylase in the cytoplasm. Curr. Biol. 2000, 10, 747–749. [Google Scholar] [CrossRef]
- Boyault, C.; Gilquin, B.; Zhang, Y.; Rybin, V.; Garman, E.; Meyer-Klaucke, W.; Matthias, P.; Müller, C.W.; Khochbin, S. HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J. 2006, 25, 3357–3366. [Google Scholar] [CrossRef] [PubMed]
- Seigneurin-Berny, D.; Verdel, A.; Curtet, S.; Lemercier, C.; Garin, J.; Rousseaux, S.; Khochbin, S. Identification of components of the murine histone deacetylase 6 complex: Link between acetylation and ubiquitination signaling pathways. Mol. Cell. Biol. 2001, 21, 8035–8044. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gilquin, B.; Khochbin, S.; Matthias, P. Two catalytic domains are required for protein deacetylation. J. Biol. Chem. 2006, 281, 2401–2404. [Google Scholar] [CrossRef] [PubMed]
- Hai, Y.; Christianson, D.W. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol. 2016, 12, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.-W.; Shin, D.-H.; Lee, D.H.; Choi, J.; Han, G.; Lee, K.Y.; Kwon, S.H. HDAC6 deacetylates p53 at lysines 381/382 and differentially coordinates p53-induced apoptosis. Cancer Lett. 2017, 391, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xiang, S.; Joo, H.-Y.; Wang, L.; Williams, K.A.; Liu, W.; Hu, C.; Tong, D.; Haakenson, J.; Wang, C.; et al. HDAC6 deacetylates and ubiquitinates MSH2 to maintain proper levels of MutSα. Mol. Cell 2014, 55, 31–46. [Google Scholar] [CrossRef]
- Casero, R.A.; Murray Stewart, T.; Pegg, A.E. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 2018, 18, 681–695. [Google Scholar] [CrossRef]
- Hai, Y.; Shinsky, S.A.; Porter, N.J.; Christianson, D.W. Histone deacetylase 10 structure and molecular function as a polyamine deacetylase. Nat. Commun. 2017, 8, 15368. [Google Scholar] [CrossRef]
- Oehme, I.; Linke, J.-P.; Böck, B.C.; Milde, T.; Lodrini, M.; Hartenstein, B.; Wiegand, I.; Eckert, C.; Roth, W.; Kool, M.; et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc. Natl. Acad. Sci. USA 2013, 110, E2592–E2601. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Li, Y.; Xiang, S.; Yuan, F.; Yuan, Z.; Telles, E.; Fang, J.; Coppola, D.; Shibata, D.; Lane, W.S.; et al. Histone deacetylase 10 regulates DNA mismatch repair and may involve the deacetylation of MutS homolog 2. J. Biol. Chem. 2015, 290, 22795–22804. [Google Scholar] [CrossRef]
- Moreno-Yruela, C.; Galleano, I.; Madsen, A.S.; Olsen, C.A. Histone Deacetylase 11 Is an ε-N-Myristoyllysine Hydrolase. Cell Chem. Biol. 2018, 25, 849–856. [Google Scholar] [CrossRef]
- Kutil, Z.; Novakova, Z.; Meleshin, M.; Mikesova, J.; Schutkowski, M.; Barinka, C. Histone Deacetylase 11 Is a Fatty-Acid Deacylase. ACS Chem. Biol. 2018, 13, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Sun, L.; Aramsangtienchai, P.; Spiegelman, N.A.; Zhang, X.; Huang, W.; Seto, E.; Lin, H. HDAC11 regulates type I interferon signaling through defatty-acylation of SHMT2. Proc. Natl. Acad. Sci. USA 2019, 116, 5487–5492. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium Human Genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Colón-Díaz, M.; Báez-Vega, P.; García, M.; Ruiz, A.; Monteiro, J.B.; Fourquet, J.; Bayona, M.; Alvarez-Garriga, C.; Achille, A.; Seto, E.; et al. HDAC1 and HDAC2 are differentially expressed in endometriosis. Reprod. Sci. 2012, 19, 483–492. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Jawerka, M.; Colak, D.; Dimou, L.; Spiller, C.; Lagger, S.; Montgomery, R.L.; Olson, E.N.; Wurst, W.; Göttlicher, M.; Götz, M. The specific role of histone deacetylase 2 in adult neurogenesis. Neuron Glia Biol. 2010, 6, 93–107. [Google Scholar] [CrossRef]
- Reichert, N.; Choukrallah, M.-A.; Matthias, P. Multiple roles of class I HDACs in proliferation, differentiation, and development. Cell. Mol. Life Sci. 2012, 69, 2173–2187. [Google Scholar] [CrossRef]
- Grausenburger, R.; Bilic, I.; Boucheron, N.; Zupkovitz, G.; El-Housseiny, L.; Tschismarov, R.; Zhang, Y.; Rembold, M.; Gaisberger, M.; Hartl, A.; et al. Conditional deletion of histone deacetylase 1 in T cells leads to enhanced airway inflammation and increased Th2 cytokine production. J. Immunol. 2010, 185, 3489–3497. [Google Scholar] [CrossRef]
- Emmett, M.J.; Lazar, M.A. Integrative regulation of physiology by histone deacetylase 3. Nat. Rev. Mol. Cell Biol. 2019, 20, 102–115. [Google Scholar] [CrossRef]
- Li, J.; Chen, S.; Cleary, R.A.; Wang, R.; Gannon, O.J.; Seto, E.; Tang, D.D. Histone deacetylase 8 regulates cortactin deacetylation and contraction in smooth muscle tissues. Am. J. Physiol. Cell Physiol. 2014, 307, C288–C295. [Google Scholar] [CrossRef] [PubMed]
- Parra, M. Class IIa HDACs - new insights into their functions in physiology and pathology. FEBS J. 2015, 282, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- Seidel, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Histone deacetylase 6 in health and disease. Epigenomics 2015, 7, 103–118. [Google Scholar] [CrossRef]
- Iaconelli, J.; Xuan, L.; Karmacharya, R. HDAC6 Modulates Signaling Pathways Relevant to Synaptic Biology and Neuronal Differentiation in Human Stem-Cell-Derived Neurons. Int. J. Mol. Sci. 2019, 20, 1605. [Google Scholar] [CrossRef]
- Dahiya, S.; Beier, U.H.; Wang, L.; Han, R.; Jiao, J.; Akimova, T.; Angelin, A.; Wallace, D.C.; Hancock, W.W. HDAC10 deletion promotes Foxp3+ T-regulatory cell function. Sci. Rep. 2020, 10, 424. [Google Scholar] [CrossRef] [PubMed]
- Yanginlar, C.; Logie, C. HDAC11 is a regulator of diverse immune functions. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 54–59. [Google Scholar] [CrossRef]
- Bantscheff, M.; Hopf, C.; Savitski, M.M.; Dittmann, A.; Grandi, P.; Michon, A.-M.; Schlegl, J.; Abraham, Y.; Becher, I.; Bergamini, G.; et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 2011, 29, 255–265. [Google Scholar] [CrossRef]
- Adams, G.E.; Chandru, A.; Cowley, S.M. Co-repressor, co-activator and general transcription factor: The many faces of the Sin3 histone deacetylase (HDAC) complex. Biochem. J. 2018, 475, 3921–3932. [Google Scholar] [CrossRef]
- Bansal, N.; David, G.; Farias, E.; Waxman, S. Emerging Roles of Epigenetic Regulator Sin3 in Cancer. Adv. Cancer Res. 2016, 130, 113–135. [Google Scholar] [CrossRef]
- Schreiber-Agus, N.; Chin, L.; Chen, K.; Torres, R.; Rao, G.; Guida, P.; Skoultchi, A.I.; DePinho, R.A. An amino-terminal domain of Mxi1 mediates anti-Myc oncogenic activity and interacts with a homolog of the yeast transcriptional repressor SIN3. Cell 1995, 80, 777–786. [Google Scholar] [CrossRef]
- Lai, A.; Kennedy, B.K.; Barbie, D.A.; Bertos, N.R.; Yang, X.J.; Theberge, M.C.; Tsai, S.C.; Seto, E.; Zhang, Y.; Kuzmichev, A.; et al. RBP1 recruits the mSIN3-histone deacetylase complex to the pocket of retinoblastoma tumor suppressor family proteins found in limited discrete regions of the nucleus at growth arrest. Mol. Cell. Biol. 2001, 21, 2918–2932. [Google Scholar] [CrossRef]
- Laherty, C.D.; Yang, W.M.; Sun, J.M.; Davie, J.R.; Seto, E.; Eisenman, R.N. Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell 1997, 89, 349–356. [Google Scholar] [CrossRef]
- Hassig, C.A.; Fleischer, T.C.; Billin, A.N.; Schreiber, S.L.; Ayer, D.E. Histone deacetylase activity is required for full transcriptional repression by mSin3A. Cell 1997, 89, 341–347. [Google Scholar] [CrossRef]
- Pellegrino, J.; Castrillon, D.H.; David, G. Chromatin associated Sin3A is essential for male germ cell lineage in the mouse. Dev. Biol. 2012, 369, 349–355. [Google Scholar] [CrossRef][Green Version]
- Wang, Y.; Tian, Y.; Morley, M.P.; Lu, M.M.; Demayo, F.J.; Olson, E.N.; Morrisey, E.E. Development and regeneration of Sox2+ endoderm progenitors are regulated by a Hdac1/2-Bmp4/Rb1 regulatory pathway. Dev. Cell 2013, 24, 345–358. [Google Scholar] [CrossRef]
- Yao, C.; Carraro, G.; Konda, B.; Guan, X.; Mizuno, T.; Chiba, N.; Kostelny, M.; Kurkciyan, A.; David, G.; McQualter, J.L.; et al. Sin3a regulates epithelial progenitor cell fate during lung development. Development 2017, 144, 2618–2628. [Google Scholar] [CrossRef]
- Lin, R.J.; Nagy, L.; Inoue, S.; Shao, W.; Miller, W.H.; Evans, R.M. Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature 1998, 391, 811–814. [Google Scholar] [CrossRef]
- Guruharsha, K.G.; Rual, J.-F.; Zhai, B.; Mintseris, J.; Vaidya, P.; Vaidya, N.; Beekman, C.; Wong, C.; Rhee, D.Y.; Cenaj, O.; et al. A protein complex network of Drosophila melanogaster. Cell 2011, 147, 690–703. [Google Scholar] [CrossRef]
- Das, T.K.; Sangodkar, J.; Negre, N.; Narla, G.; Cagan, R.L. Sin3a acts through a multi-gene module to regulate invasion in Drosophila and human tumors. Oncogene 2013, 32, 3184–3197. [Google Scholar] [CrossRef]
- DiMauro, T.; Cantor, D.J.; Bainor, A.J.; David, G. Transcriptional repression of Sin3B by Bmi-1 prevents cellular senescence and is relieved by oncogene activation. Oncogene 2015, 34, 4011–4017. [Google Scholar] [CrossRef]
- Ling, J.; Kang, Y.; Zhao, R.; Xia, Q.; Lee, D.-F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.B.; Wang, H.; et al. KrasG12D-induced IKK2/β/NF-κB activation by IL-1α and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 105–120. [Google Scholar] [CrossRef]
- Lakowski, B.; Roelens, I.; Jacob, S. CoREST-like complexes regulate chromatin modification and neuronal gene expression. J. Mol. Neurosci. 2006, 29, 227–239. [Google Scholar] [CrossRef]
- Laugesen, A.; Helin, K. Chromatin repressive complexes in stem cells, development, and cancer. Cell Stem Cell 2014, 14, 735–751. [Google Scholar] [CrossRef]
- Song, Y.; Dagil, L.; Fairall, L.; Robertson, N.; Wu, M.; Ragan, T.J.; Savva, C.G.; Saleh, A.; Morone, N.; Kunze, M.B.A.; et al. Mechanism of Crosstalk between the LSD1 Demethylase and HDAC1 Deacetylase in the CoREST Complex. Cell Rep. 2020, 30, 2699–2711. [Google Scholar] [CrossRef]
- Hayakawa, T.; Nakayama, J.-I. Physiological roles of class I HDAC complex and histone demethylase. J. Biomed. Biotechnol. 2011, 2011, 129383. [Google Scholar] [CrossRef]
- You, A.; Tong, J.K.; Grozinger, C.M.; Schreiber, S.L. CoREST is an integral component of the CoREST- human histone deacetylase complex. Proc. Natl. Acad. Sci. USA 2001, 98, 1454–1458. [Google Scholar] [CrossRef]
- Shi, Y.-J.; Matson, C.; Lan, F.; Iwase, S.; Baba, T.; Shi, Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol. Cell 2005, 19, 857–864. [Google Scholar] [CrossRef]
- Pilotto, S.; Speranzini, V.; Tortorici, M.; Durand, D.; Fish, A.; Valente, S.; Forneris, F.; Mai, A.; Sixma, T.K.; Vachette, P.; et al. Interplay among nucleosomal DNA, histone tails, and corepressor CoREST underlies LSD1-mediated H3 demethylation. Proc. Natl. Acad. Sci. USA 2015, 112, 2752–2757. [Google Scholar] [CrossRef]
- Kooistra, S.M.; Helin, K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 2012, 13, 297–311. [Google Scholar] [CrossRef]
- Hakimi, M.-A.; Bochar, D.A.; Chenoweth, J.; Lane, W.S.; Mandel, G.; Shiekhattar, R. A core-BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes. Proc. Natl. Acad. Sci. USA 2002, 99, 7420–7425. [Google Scholar] [CrossRef]
- Lee, M.G.; Wynder, C.; Cooch, N.; Shiekhattar, R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 2005, 437, 432–435. [Google Scholar] [CrossRef]
- Sumoy, L.; Carim, L.; Escarceller, M.; Nadal, M.; Gratacòs, M.; Pujana, M.A.; Estivill, X.; Peral, B. HMG20A and HMG20B map to human chromosomes 15q24 and 19p13.3 and constitute a distinct class of HMG-box genes with ubiquitous expression. Cytogenet. Cell Genet. 2000, 88, 62–67. [Google Scholar] [CrossRef]
- McClellan, D.; Casey, M.J.; Bareyan, D.; Lucente, H.; Ours, C.; Velinder, M.; Singer, J.; Lone, M.D.; Sun, W.; Coria, Y.; et al. Growth Factor Independence 1B-Mediated Transcriptional Repression and Lineage Allocation Require Lysine-Specific Demethylase 1-Dependent Recruitment of the BHC Complex. Mol. Cell. Biol. 2019, 39. [Google Scholar] [CrossRef]
- Zalloum, W.A.; Zalloum, H.M. Exploring the Active Center of the LSD1/CoREST Complex by Molecular Dynamics Simulation Utilizing Its Co-crystallized Co-factor Tetrahydrofolate as a Probe. J. Chem. Inf. Model. 2017, 57, 3022–3031. [Google Scholar] [CrossRef]
- de Castro, I.J.; Amin, H.A.; Vinciotti, V.; Vagnarelli, P. Network of phosphatases and HDAC complexes at repressed chromatin. Cell Cycle 2017, 16, 2011–2017. [Google Scholar] [CrossRef]
- Saleque, S.; Kim, J.; Rooke, H.M.; Orkin, S.H. Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol. Cell 2007, 27, 562–572. [Google Scholar] [CrossRef]
- van Bergen, M.G.J.M.; van der Reijden, B.A. Targeting the GFI1/1B-CoREST Complex in Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 1027. [Google Scholar] [CrossRef]
- Torchy, M.P.; Hamiche, A.; Klaholz, B.P. Structure and function insights into the NuRD chromatin remodeling complex. Cell. Mol. Life Sci. 2015, 72, 2491–2507. [Google Scholar] [CrossRef]
- Torrado, M.; Low, J.K.K.; Silva, A.P.G.; Schmidberger, J.W.; Sana, M.; Tabar, M.S.; Isilak, M.E.; Winning, C.S.; Kwong, C.; Bedward, M.J.; et al. Refinement of the subunit interaction network within the nucleosome remodelling and deacetylase (NuRD) complex. FEBS J. 2017, 284, 4216–4232. [Google Scholar] [CrossRef]
- Zhang, T.; Wei, G.; Millard, C.J.; Fischer, R.; Konietzny, R.; Kessler, B.M.; Schwabe, J.W.R.; Brockdorff, N. A variant NuRD complex containing PWWP2A/B excludes MBD2/3 to regulate transcription at active genes. Nat. Commun. 2018, 9, 3798. [Google Scholar] [CrossRef]
- Brero, A.; Easwaran, H.P.; Nowak, D.; Grunewald, I.; Cremer, T.; Leonhardt, H.; Cardoso, M.C. Methyl CpG-binding proteins induce large-scale chromatin reorganization during terminal differentiation. J. Cell Biol. 2005, 169, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, O.; Li, R.; Hung, J.-H.; Chen, P.B.; Dong, X.; Ee, L.-S.; Weng, Z.; Rando, O.J.; Fazzio, T.G. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell 2011, 147, 1498–1510. [Google Scholar] [CrossRef] [PubMed]
- Basta, J.; Rauchman, M. The nucleosome remodeling and deacetylase complex in development and disease. Transl. Res. 2015, 165, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Millard, C.J.; Varma, N.; Saleh, A.; Morris, K.; Watson, P.J.; Bottrill, A.R.; Fairall, L.; Smith, C.J.; Schwabe, J.W.R. The structure of the core NuRD repression complex provides insights into its interaction with chromatin. Elife 2016, 5, e13941. [Google Scholar] [CrossRef]
- Mor, N.; Rais, Y.; Sheban, D.; Peles, S.; Aguilera-Castrejon, A.; Zviran, A.; Elinger, D.; Viukov, S.; Geula, S.; Krupalnik, V.; et al. Neutralizing Gatad2a-Chd4-Mbd3/NuRD Complex Facilitates Deterministic Induction of Naive Pluripotency. Cell Stem Cell 2018, 23, 412–425. [Google Scholar] [CrossRef]
- Shao, S.; Cao, H.; Wang, Z.; Zhou, D.; Wu, C.; Wang, S.; Xia, D.; Zhang, D. CHD4/NuRD complex regulates complement gene expression and correlates with CD8 T cell infiltration in human hepatocellular carcinoma. Clin. Epigenetics 2020, 12, 31. [Google Scholar] [CrossRef]
- Nio, K.; Yamashita, T.; Okada, H.; Kondo, M.; Hayashi, T.; Hara, Y.; Nomura, Y.; Zeng, S.S.; Yoshida, M.; Hayashi, T.; et al. Defeating EpCAM(+) liver cancer stem cells by targeting chromatin remodeling enzyme CHD4 in human hepatocellular carcinoma. J. Hepatol. 2015, 63, 1164–1172. [Google Scholar] [CrossRef]
- Pagliuca, F.W.; Collins, M.O.; Lichawska, A.; Zegerman, P.; Choudhary, J.S.; Pines, J. Quantitative proteomics reveals the basis for the biochemical specificity of the cell-cycle machinery. Mol. Cell 2011, 43, 406–417. [Google Scholar] [CrossRef]
- Millard, C.J.; Watson, P.J.; Fairall, L.; Schwabe, J.W.R. Targeting Class I Histone Deacetylases in a “Complex” Environment. Trends Pharmacol. Sci. 2017, 38, 363–377. [Google Scholar] [CrossRef]
- Ordentlich, P.; Downes, M.; Xie, W.; Genin, A.; Spinner, N.B.; Evans, R.M. Unique forms of human and mouse nuclear receptor corepressor SMRT. Proc. Natl. Acad. Sci. USA 1999, 96, 2639–2644. [Google Scholar] [CrossRef]
- Shimizu, H.; Lu, Y.; Vella, K.R.; Damilano, F.; Astapova, I.; Amano, I.; Ritter, M.; Gallop, M.R.; Rosenzweig, A.N.; Cohen, R.N.; et al. Nuclear corepressor SMRT is a strong regulator of body weight independently of its ability to regulate thyroid hormone action. PLoS ONE 2019, 14, e0220717. [Google Scholar] [CrossRef] [PubMed]
- Perissi, V.; Jepsen, K.; Glass, C.K.; Rosenfeld, M.G. Deconstructing repression: Evolving models of co-repressor action. Nat. Rev. Genet. 2010, 11, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Liang, N.; Jakobsson, T.; Fan, R.; Treuter, E. The Nuclear Receptor-Co-repressor Complex in Control of Liver Metabolism and Disease. Front. Endocrinol. (Lausanne) 2019, 10, 411. [Google Scholar] [CrossRef] [PubMed]
- Stengel, K.R.; Hiebert, S.W. Class I HDACs Affect DNA Replication, Repair, and Chromatin Structure: Implications for Cancer Therapy. Antioxid. Redox Signal. 2015, 23, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Soufi, A.; Donahue, G.; Zaret, K.S. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 2012, 151, 994–1004. [Google Scholar] [CrossRef]
- Zhuang, Q.; Li, W.; Benda, C.; Huang, Z.; Ahmed, T.; Liu, P.; Guo, X.; Ibañez, D.P.; Luo, Z.; Zhang, M.; et al. NCoR/SMRT co-repressors cooperate with c-MYC to create an epigenetic barrier to somatic cell reprogramming. Nat. Cell Biol. 2018, 20, 400–412. [Google Scholar] [CrossRef]
- Mottis, A.; Mouchiroud, L.; Auwerx, J. Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev. 2013, 27, 819–835. [Google Scholar] [CrossRef]
- Stengel, K.R.; Barnett, K.R.; Wang, J.; Liu, Q.; Hodges, E.; Hiebert, S.W.; Bhaskara, S. Deacetylase activity of histone deacetylase 3 is required for productive VDJ recombination and B-cell development. Proc. Natl. Acad. Sci. USA 2017, 114, 8608–8613. [Google Scholar] [CrossRef]
- You, S.-H.; Lim, H.-W.; Sun, Z.; Broache, M.; Won, K.-J.; Lazar, M.A. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat. Struct. Mol. Biol. 2013, 20, 182–187. [Google Scholar] [CrossRef]
- Wong, M.M.; Guo, C.; Zhang, J. Nuclear receptor corepressor complexes in cancer: Mechanism, function and regulation. Am. J. Clin. Exp. Urol. 2014, 2, 169–187. [Google Scholar]
- Hong, S.H.; David, G.; Wong, C.W.; Dejean, A.; Privalsky, M.L. SMRT corepressor interacts with PLZF and with the PML-retinoic acid receptor alpha (RARalpha) and PLZF-RARalpha oncoproteins associated with acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 1997, 94, 9028–9033. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, A.; Tomita, A.; Kiyoi, H.; Naoe, T. Histone deacetylase 3 (HDAC3) is recruited to target promoters by PML-RARalpha as a component of the N-CoR co-repressor complex to repress transcription in vivo. Biochem. Biophys. Res. Commun. 2006, 345, 1471–1480. [Google Scholar] [CrossRef] [PubMed]
- Racanicchi, S.; Maccherani, C.; Liberatore, C.; Billi, M.; Gelmetti, V.; Panigada, M.; Rizzo, G.; Nervi, C.; Grignani, F. Targeting fusion protein/corepressor contact restores differentiation response in leukemia cells. EMBO J. 2005, 24, 1232–1242. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Qin, G.; Zhao, T.C. HDAC4: Mechanism of regulation and biological functions. Epigenomics 2014, 6, 139–150. [Google Scholar] [CrossRef]
- Park, S.-Y.; Kim, G.S.; Hwang, H.-J.; Nam, T.-H.; Park, H.-S.; Song, J.; Jang, T.-H.; Lee, Y.C.; Kim, J.-S. Structural basis of the specific interaction of SMRT corepressor with histone deacetylase 4. Nucleic Acids Res. 2018, 46, 11776–11788. [Google Scholar] [CrossRef]
- Kim, M.-S.; Akhtar, M.W.; Adachi, M.; Mahgoub, M.; Bassel-Duby, R.; Kavalali, E.T.; Olson, E.N.; Monteggia, L.M. An essential role for histone deacetylase 4 in synaptic plasticity and memory formation. J. Neurosci. 2012, 32, 10879–10886. [Google Scholar] [CrossRef]
- Sando, R.; Gounko, N.; Pieraut, S.; Liao, L.; Yates, J.; Maximov, A. HDAC4 governs a transcriptional program essential for synaptic plasticity and memory. Cell 2012, 151, 821–834. [Google Scholar] [CrossRef]
- Williams, S.R.; Aldred, M.A.; Der Kaloustian, V.M.; Halal, F.; Gowans, G.; McLeod, D.R.; Zondag, S.; Toriello, H.V.; Magenis, R.E.; Elsea, S.H. Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. Am. J. Hum. Genet. 2010, 87, 219–228. [Google Scholar] [CrossRef]
- Li, J.; Chen, J.; Ricupero, C.L.; Hart, R.P.; Schwartz, M.S.; Kusnecov, A.; Herrup, K. Nuclear accumulation of HDAC4 in ATM deficiency promotes neurodegeneration in ataxia telangiectasia. Nat. Med. 2012, 18, 783–790. [Google Scholar] [CrossRef]
- Hassell, K.N. Histone Deacetylases and their Inhibitors in Cancer Epigenetics. Diseases 2019, 7, 57. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, L.; Dahiya, S.; Beier, U.H.; Han, R.; Samanta, A.; Bergman, J.; Sotomayor, E.M.; Seto, E.; Kozikowski, A.P.; et al. Histone/protein deacetylase 11 targeting promotes Foxp3+ Treg function. Sci. Rep. 2017, 7, 8626. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Greco, T.M.; Guise, A.J.; Luo, Y.; Yu, F.; Nesvizhskii, A.I.; Cristea, I.M. The functional interactome landscape of the human histone deacetylase family. Mol. Syst. Biol. 2013, 9, 672. [Google Scholar] [CrossRef]
- Gubitz, A.K.; Feng, W.; Dreyfuss, G. The SMN complex. Exp. Cell Res. 2004, 296, 51–56. [Google Scholar] [CrossRef]
- Saxena, S.; Yuan, P.; Dhar, S.K.; Senga, T.; Takeda, D.; Robinson, H.; Kornbluth, S.; Swaminathan, K.; Dutta, A. A dimerized coiled-coil domain and an adjoining part of geminin interact with two sites on Cdt1 for replication inhibition. Mol. Cell 2004, 15, 245–258. [Google Scholar] [CrossRef]
- Glozak, M.A.; Seto, E. Acetylation/deacetylation modulates the stability of DNA replication licensing factor Cdt1. J. Biol. Chem. 2009, 284, 11446–11453. [Google Scholar] [CrossRef] [PubMed]
- Mercatelli, D.; Ray, F.; Giorgi, F.M. Pan-Cancer and Single-Cell Modeling of Genomic Alterations Through Gene Expression. Front. Genet. 2019, 10, 671. [Google Scholar] [CrossRef] [PubMed]
- Rajbhandari, P.; Lopez, G.; Capdevila, C.; Salvatori, B.; Yu, J.; Rodriguez-Barrueco, R.; Martinez, D.; Yarmarkovich, M.; Weichert-Leahey, N.; Abraham, B.J.; et al. Cross-Cohort Analysis Identifies a TEAD4-MYCN Positive Feedback Loop as the Core Regulatory Element of High-Risk Neuroblastoma. Cancer Discov. 2018, 8, 582–599. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef]
- Liu, X.; Yu, Y.; Zhang, J.; Lu, C.; Wang, L.; Liu, P.; Song, H. HDAC1 Silencing in Ovarian Cancer Enhances the Chemotherapy Response. Cell. Physiol. Biochem. 2018, 48, 1505–1518. [Google Scholar] [CrossRef]
- Zhang, L.; Bu, L.; Hu, J.; Xu, Z.; Ruan, L.; Fang, Y.; Wang, P. HDAC1 knockdown inhibits invasion and induces apoptosis in non-small cell lung cancer cells. Biol. Chem. 2018, 399, 603–610. [Google Scholar] [CrossRef]
- Seo, J.; Min, S.K.; Park, H.-R.; Kim, D.H.; Kwon, M.J.; Kim, L.S.; Ju, Y.-S. Expression of Histone Deacetylases HDAC1, HDAC2, HDAC3, and HDAC6 in Invasive Ductal Carcinomas of the Breast. J. Breast Cancer 2014, 17, 323–331. [Google Scholar] [CrossRef]
- Stubbs, M.C.; Kim, W.; Bariteau, M.; Davis, T.; Vempati, S.; Minehart, J.; Witkin, M.; Qi, J.; Krivtsov, A.V.; Bradner, J.E.; et al. Selective Inhibition of HDAC1 and HDAC2 as a Potential Therapeutic Option for B-ALL. Clin. Cancer Res. 2015, 21, 2348–2358. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-H.; Seo, D.; Choi, K.-J.; Andersen, J.B.; Won, M.-A.; Kitade, M.; Gómez-Quiroz, L.E.; Judge, A.D.; Marquardt, J.U.; Raggi, C.; et al. Antitumor effects in hepatocarcinoma of isoform-selective inhibition of HDAC2. Cancer Res. 2014, 74, 4752–4761. [Google Scholar] [CrossRef] [PubMed]
- Frumm, S.M.; Fan, Z.P.; Ross, K.N.; Duvall, J.R.; Gupta, S.; VerPlank, L.; Suh, B.-C.; Holson, E.; Wagner, F.F.; Smith, W.B.; et al. Selective HDAC1/HDAC2 inhibitors induce neuroblastoma differentiation. Chem. Biol. 2013, 20, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Coni, S.; Mancuso, A.B.; Di Magno, L.; Sdruscia, G.; Manni, S.; Serrao, S.M.; Rotili, D.; Spiombi, E.; Bufalieri, F.; Petroni, M.; et al. Selective targeting of HDAC1/2 elicits anticancer effects through Gli1 acetylation in preclinical models of SHH Medulloblastoma. Sci. Rep. 2017, 7, 44079. [Google Scholar] [CrossRef]
- McLeod, A.B.; Stice, J.P.; Wardell, S.E.; Alley, H.M.; Chang, C.-Y.; McDonnell, D.P. Validation of histone deacetylase 3 as a therapeutic target in castration-resistant prostate cancer. Prostate 2018, 78, 266–277. [Google Scholar] [CrossRef]
- Mondello, P.; Tadros, S.; Teater, M.; Fontan, L.; Chang, A.Y.; Jain, N.; Yang, H.; Singh, S.; Ying, H.-Y.; Chu, C.-S.; et al. Selective Inhibition of HDAC3 Targets Synthetic Vulnerabilities and Activates Immune Surveillance in Lymphoma. Cancer Discov. 2020, 10, 440–459. [Google Scholar] [CrossRef]
- Hsieh, H.-Y.; Chuang, H.-C.; Shen, F.-H.; Detroja, K.; Hsin, L.-W.; Chen, C.-S. Targeting breast cancer stem cells by novel HDAC3-selective inhibitors. Eur. J. Med. Chem. 2017, 140, 42–51. [Google Scholar] [CrossRef]
- Rettig, I.; Koeneke, E.; Trippel, F.; Mueller, W.C.; Burhenne, J.; Kopp-Schneider, A.; Fabian, J.; Schober, A.; Fernekorn, U.; von Deimling, A.; et al. Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid-mediated differentiation. Cell Death Dis. 2015, 6, e1657. [Google Scholar] [CrossRef]
- Li, A.; Liu, Z.; Li, M.; Zhou, S.; Xu, Y.; Xiao, Y.; Yang, W. HDAC5, a potential therapeutic target and prognostic biomarker, promotes proliferation, invasion and migration in human breast cancer. Oncotarget 2016, 7, 37966–37978. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Cao, F.; Yu, X.; Zhou, P.; Di, Q.; Lei, J.; Tai, Y.; Wu, H.; Li, X.; Wang, X.; et al. The expression of HDAC7 in cancerous gastric tissues is positively associated with distant metastasis and poor patient prognosis. Clin. Transl. Oncol. 2017, 19, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Yano, M.; Yasuda, M.; Sakaki, M.; Nagata, K.; Fujino, T.; Arai, E.; Hasebe, T.; Miyazawa, M.; Miyazawa, M.; Ogane, N.; et al. Association of histone deacetylase expression with histology and prognosis of ovarian cancer. Oncol. Lett. 2018, 15, 3524–3531. [Google Scholar] [CrossRef]
- Li, T.; Zhang, C.; Hassan, S.; Liu, X.; Song, F.; Chen, K.; Zhang, W.; Yang, J. Histone deacetylase 6 in cancer. J. Hematol. Oncol. 2018, 11, 111. [Google Scholar] [CrossRef] [PubMed]
- Depetter, Y.; Geurs, S.; De Vreese, R.; Goethals, S.; Vandoorn, E.; Laevens, A.; Steenbrugge, J.; Meyer, E.; de Tullio, P.; Bracke, M.; et al. Selective pharmacological inhibitors of HDAC6 reveal biochemical activity but functional tolerance in cancer models. Int. J. Cancer 2019, 145, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Thole, T.M.; Lodrini, M.; Fabian, J.; Wuenschel, J.; Pfeil, S.; Hielscher, T.; Kopp-Schneider, A.; Heinicke, U.; Fulda, S.; Witt, O.; et al. Neuroblastoma cells depend on HDAC11 for mitotic cell cycle progression and survival. Cell Death Dis. 2017, 8, e2635. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef]
- Benedetti, R.; Conte, M.; Altucci, L. Targeting Histone Deacetylases in Diseases: Where Are We? Antioxid. Redox Signal. 2015, 23, 99–126. [Google Scholar] [CrossRef]
- Melesina, J.; Robaa, D.; Pierce, R.J.; Romier, C.; Sippl, W. Homology modeling of parasite histone deacetylases to guide the structure-based design of selective inhibitors. J. Mol. Graph. Model. 2015, 62, 342–361. [Google Scholar] [CrossRef]
- Stockhausen, M.-T.; Sjölund, J.; Manetopoulos, C.; Axelson, H. Effects of the histone deacetylase inhibitor valproic acid on Notch signalling in human neuroblastoma cells. Br. J. Cancer 2005, 92, 751–759. [Google Scholar] [CrossRef]
- Riggs, M.G.; Whittaker, R.G.; Neumann, J.R.; Ingram, V.M. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature 1977, 268, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M.; Emiliani, S.; Verdin, E.; Webb, Y.; Breslow, R.; Rifkind, R.A.; Marks, P.A. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc. Natl. Acad. Sci. USA 1998, 95, 3003–3007. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, N.; Kobayashi, M.; Nagashima, K.; Wakisaka, Y.; Koizumi, K. A new antifungal antibiotic, trichostatin. J. Antibiot. 1976, 29, 1–6. [Google Scholar] [CrossRef]
- Banik, D.; Moufarrij, S.; Villagra, A. Immunoepigenetics Combination Therapies: An Overview of the Role of HDACs in Cancer Immunotherapy. Int J. Mol. Sci. 2019, 20, 2241. [Google Scholar] [CrossRef]
- Rahmani, M.; Aust, M.M.; Benson, E.C.; Wallace, L.; Friedberg, J.; Grant, S. PI3K/mTOR inhibition markedly potentiates HDAC inhibitor activity in NHL cells through BIM- and MCL-1-dependent mechanisms in vitro and in vivo. Clin. Cancer Res. 2014, 20, 4849–4860. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Shi, W.; Jankowski, K.; Kan, C.-Y.; Cluse, L.; Martin, B.P.; MacKenzie, K.L.; Smyth, G.K.; Johnstone, R.W. HDAC inhibitors induce tumor-cell-selective pro-apoptotic transcriptional responses. Cell Death Dis. 2013, 4, e519. [Google Scholar] [CrossRef]
- Rada-Iglesias, A.; Enroth, S.; Ameur, A.; Koch, C.M.; Clelland, G.K.; Respuela-Alonso, P.; Wilcox, S.; Dovey, O.M.; Ellis, P.D.; Langford, C.F.; et al. Butyrate mediates decrease of histone acetylation centered on transcription start sites and down-regulation of associated genes. Genome Res. 2007, 17, 708–719. [Google Scholar] [CrossRef]
- Krämer, O.H.; Göttlicher, M.; Heinzel, T. Histone deacetylase as a therapeutic target. Trends Endocrinol. Metab. 2001, 12, 294–300. [Google Scholar] [CrossRef]
- Van Lint, C.; Emiliani, S.; Verdin, E. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr. 1996, 5, 245–253. [Google Scholar]
- Fukumoto, T.; Park, P.H.; Wu, S.; Fatkhutdinov, N.; Karakashev, S.; Nacarelli, T.; Kossenkov, A.V.; Speicher, D.W.; Jean, S.; Zhang, L.; et al. Repurposing Pan-HDAC Inhibitors for ARID1A-Mutated Ovarian Cancer. Cell Rep. 2018, 22, 3393–3400. [Google Scholar] [CrossRef]
- Jia, D.; Augert, A.; Kim, D.-W.; Eastwood, E.; Wu, N.; Ibrahim, A.H.; Kim, K.-B.; Dunn, C.T.; Pillai, S.P.S.; Gazdar, A.F.; et al. Crebbp Loss Drives Small Cell Lung Cancer and Increases Sensitivity to HDAC Inhibition. Cancer Discov. 2018, 8, 1422–1437. [Google Scholar] [CrossRef] [PubMed]
- Garrido Castro, P.; van Roon, E.H.J.; Pinhanços, S.S.; Trentin, L.; Schneider, P.; Kerstjens, M.; Te Kronnie, G.; Heidenreich, O.; Pieters, R.; Stam, R.W. The HDAC inhibitor panobinostat (LBH589) exerts in vivo anti-leukaemic activity against MLL-rearranged acute lymphoblastic leukaemia and involves the RNF20/RNF40/WAC-H2B ubiquitination axis. Leukemia 2018, 32, 323–331. [Google Scholar] [CrossRef]
- Lodrini, M.; Oehme, I.; Schroeder, C.; Milde, T.; Schier, M.C.; Kopp-Schneider, A.; Schulte, J.H.; Fischer, M.; De Preter, K.; Pattyn, F.; et al. MYCN and HDAC2 cooperate to repress miR-183 signaling in neuroblastoma. Nucleic Acids Res. 2013, 41, 6018–6033. [Google Scholar] [CrossRef]
- Agarwal, S.; Milazzo, G.; Rajapakshe, K.; Bernardi, R.; Chen, Z.; Barbieri, E.; Koster, J.; Perini, G.; Coarfa, C.; Shohet, J.M. MYCN acts as a direct co-regulator of p53 in MYCN amplified neuroblastoma. Oncotarget 2018, 9, 20323–20338. [Google Scholar] [CrossRef]
- Durbin, A.D.; Zimmerman, M.W.; Dharia, N.V.; Abraham, B.J.; Iniguez, A.B.; Weichert-Leahey, N.; He, S.; Krill-Burger, J.M.; Root, D.E.; Vazquez, F.; et al. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat. Genet. 2018, 50, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, J.; Liu, P.Y.; Atmadibrata, B.; Bradner, J.E.; Marshall, G.M.; Lock, R.B.; Liu, T. The Bromodomain Inhibitor JQ1 and the Histone Deacetylase Inhibitor Panobinostat Synergistically Reduce N-Myc Expression and Induce Anticancer Effects. Clin. Cancer Res. 2016, 22, 2534–2544. [Google Scholar] [CrossRef] [PubMed]
- Moffat, D.; Patel, S.; Day, F.; Belfield, A.; Donald, A.; Rowlands, M.; Wibawa, J.; Brotherton, D.; Stimson, L.; Clark, V.; et al. Discovery of 2-(6-{[(6-fluoroquinolin-2-yl)methyl]amino}bicyclo[3.1.0]hex-3-yl)-N-hydroxypyrimidine-5-carboxamide (CHR-3996), a class I selective orally active histone deacetylase inhibitor. J. Med. Chem. 2010, 53, 8663–8678. [Google Scholar] [CrossRef]
- Yee, A.J.; Bensinger, W.I.; Supko, J.G.; Voorhees, P.M.; Berdeja, J.G.; Richardson, P.G.; Libby, E.N.; Wallace, E.E.; Birrer, N.E.; Burke, J.N.; et al. Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: A multicentre phase 1b trial. Lancet Oncol. 2016, 17, 1569–1578. [Google Scholar] [CrossRef]
- Li, M.; van Esch, B.C.A.M.; Wagenaar, G.T.M.; Garssen, J.; Folkerts, G.; Henricks, P.A.J. Pro- and anti-inflammatory effects of short chain fatty acids on immune and endothelial cells. Eur. J. Pharmacol. 2018, 831, 52–59. [Google Scholar] [CrossRef]
- Chen, J.S.; Faller, D.V.; Spanjaard, R.A. Short-chain fatty acid inhibitors of histone deacetylases: Promising anticancer therapeutics? Curr. Cancer Drug Targets 2003, 3, 219–236. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal. Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.N.; Sinha, P.K. Effect of sodium butyrate on mammalian cells in culture: A review. In Vitro 1976, 12, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Sealy, L.; Chalkley, R. The effect of sodium butyrate on histone modification. Cell 1978, 14, 115–121. [Google Scholar] [CrossRef]
- Pathania, R.; Ramachandran, S.; Mariappan, G.; Thakur, P.; Shi, H.; Choi, J.-H.; Manicassamy, S.; Kolhe, R.; Prasad, P.D.; Sharma, S.; et al. Combined Inhibition of DNMT and HDAC Blocks the Tumorigenicity of Cancer Stem-like Cells and Attenuates Mammary Tumor Growth. Cancer Res. 2016, 76, 3224–3235. [Google Scholar] [CrossRef] [PubMed]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef]
- Heers, H.; Stanislaw, J.; Harrelson, J.; Lee, M.W. Valproic acid as an adjunctive therapeutic agent for the treatment of breast cancer. Eur. J. Pharmacol. 2018, 835, 61–74. [Google Scholar] [CrossRef]
- Krämer, O.H.; Zhu, P.; Ostendorff, H.P.; Golebiewski, M.; Tiefenbach, J.; Peters, M.A.; Brill, B.; Groner, B.; Bach, I.; Heinzel, T.; et al. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 2003, 22, 3411–3420. [Google Scholar] [CrossRef]
- Witt, D.; Burfeind, P.; von Hardenberg, S.; Opitz, L.; Salinas-Riester, G.; Bremmer, F.; Schweyer, S.; Thelen, P.; Neesen, J.; Kaulfuss, S. Valproic acid inhibits the proliferation of cancer cells by re-expressing cyclin D2. Carcinogenesis 2013, 34, 1115–1124. [Google Scholar] [CrossRef]
- Krumm, A.; Barckhausen, C.; Kücük, P.; Tomaszowski, K.-H.; Loquai, C.; Fahrer, J.; Krämer, O.H.; Kaina, B.; Roos, W.P. Enhanced Histone Deacetylase Activity in Malignant Melanoma Provokes RAD51 and FANCD2-Triggered Drug Resistance. Cancer Res. 2016, 76, 3067–3077. [Google Scholar] [CrossRef]
- Siu, L.L.; Pili, R.; Duran, I.; Messersmith, W.A.; Chen, E.X.; Sullivan, R.; MacLean, M.; King, S.; Brown, S.; Reid, G.K.; et al. Phase I study of MGCD0103 given as a three-times-per-week oral dose in patients with advanced solid tumors. J. Clin. Oncol. 2008, 26, 1940–1947. [Google Scholar] [CrossRef]
- Meidhof, S.; Brabletz, S.; Lehmann, W.; Preca, B.-T.; Mock, K.; Ruh, M.; Schüler, J.; Berthold, M.; Weber, A.; Burk, U.; et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol. Med. 2015, 7, 831–847. [Google Scholar] [CrossRef] [PubMed]
- Briere, D.; Sudhakar, N.; Woods, D.M.; Hallin, J.; Engstrom, L.D.; Aranda, R.; Chiang, H.; Sodré, A.L.; Olson, P.; Weber, J.S.; et al. The class I/IV HDAC inhibitor mocetinostat increases tumor antigen presentation, decreases immune suppressive cell types and augments checkpoint inhibitor therapy. Cancer Immunol. Immunother. 2018, 67, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Orillion, A.; Hashimoto, A.; Damayanti, N.; Shen, L.; Adelaiye-Ogala, R.; Arisa, S.; Chintala, S.; Ordentlich, P.; Kao, C.; Elzey, B.; et al. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 5187–5201. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.J.; McCaw, T.R.; Londono, A.I.; Katre, A.A.; Meza-Perez, S.; Yang, E.S.; Forero, A.; Buchsbaum, D.J.; Randall, T.D.; Straughn, J.M.; et al. The antitumor effects of entinostat in ovarian cancer require adaptive immunity. Cancer 2018, 124, 4657–4666. [Google Scholar] [CrossRef] [PubMed]
- Flis, S.; Gnyszka, A.; Flis, K.; Spławiński, J. MS275 enhances cytotoxicity induced by 5-fluorouracil in the colorectal cancer cells. Eur. J. Pharmacol. 2010, 627, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Hopfinger, N.R.; Nguyen, T.D.; Pogash, T.J.; Santucci-Pereira, J.; Russo, J. Epigenetic reprogramming of epithelial mesenchymal transition in triple negative breast cancer cells with DNA methyltransferase and histone deacetylase inhibitors. J. Exp. Clin. Cancer Res. 2018, 37, 314. [Google Scholar] [CrossRef]
- Choong, C.-J.; Sasaki, T.; Hayakawa, H.; Yasuda, T.; Baba, K.; Hirata, Y.; Uesato, S.; Mochizuki, H. A novel histone deacetylase 1 and 2 isoform-specific inhibitor alleviates experimental Parkinson’s disease. Neurobiol. Aging 2016, 37, 103–116. [Google Scholar] [CrossRef]
- Hirata, Y.; Sasaki, T.; Kanki, H.; Choong, C.-J.; Nishiyama, K.; Kubo, G.; Hotei, A.; Taniguchi, M.; Mochizuki, H.; Uesato, S. New 5-Aryl-Substituted 2-Aminobenzamide-Type HDAC Inhibitors with a Diketopiperazine Group and Their Ameliorating Effects on Ischemia-Induced Neuronal Cell Death. Sci. Rep. 2018, 8, 1400. [Google Scholar] [CrossRef]
- Cheng, K.; Li, S.; Liao, C. Progress in the Discovery of Macrocyclic Histone Deacetylase Inhibitors for the Treatment of Cancer. Curr. Med. Chem. 2017, 24, 4166–4179. [Google Scholar] [CrossRef]
- Maolanon, A.R.; Kristensen, H.M.E.; Leman, L.J.; Ghadiri, M.R.; Olsen, C.A. Natural and Synthetic Macrocyclic Inhibitors of the Histone Deacetylase Enzymes. Chembiochem 2017, 18, 5–49. [Google Scholar] [CrossRef]
- Mwakwari, S.C.; Patil, V.; Guerrant, W.; Oyelere, A.K. Macrocyclic histone deacetylase inhibitors. Curr. Top. Med. Chem. 2010, 10, 1423–1440. [Google Scholar] [CrossRef] [PubMed]
- Cole, K.E.; Dowling, D.P.; Boone, M.A.; Phillips, A.J.; Christianson, D.W. Structural basis of the antiproliferative activity of largazole, a depsipeptide inhibitor of the histone deacetylases. J. Am. Chem. Soc. 2011, 133, 12474–12477. [Google Scholar] [CrossRef] [PubMed]
- Cappellacci, L.; Perinelli, D.R.; Maggi, F.; Grifantini, M.; Petrelli, R. Recent Progress in Histone Deacetylase Inhibitors as Anticancer Agents. Curr. Med. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.-Y.; Wang, J.-D.; Wang, X.; Liu, H.-C.; Zhang, M.-M.; Liu, Y.-C.; Zhang, C.-H.; Su, Y.; Shen, Y.-Y.; Guo, Y.-W.; et al. Marine-derived chromopeptide A, a novel class I HDAC inhibitor, suppresses human prostate cancer cell proliferation and migration. Acta Pharmacol. Sin. 2017, 38, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.J.; Demierre, M.-F.; Kim, E.J.; Rook, A.H.; Lerner, A.; Duvic, M.; Scarisbrick, J.; Reddy, S.; Robak, T.; Becker, J.C.; et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2010, 28, 4485–4491. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.-J.; Huang, H.; He, B.; Hu, D.-H.; Li, P.-H.; Yu, Y.-J.; Zhou, X.-H.; Lv, Z.; Zhou, L.; Hu, T.-Y.; et al. Romidepsin induces G2/M phase arrest via Erk/cdc25C/cdc2/cyclinB pathway and apoptosis induction through JNK/c-Jun/caspase3 pathway in hepatocellular carcinoma cells. Biochem. Pharmacol. 2017, 127, 90–100. [Google Scholar] [CrossRef]
- Zhou, H.; Cai, Y.; Liu, D.; Li, M.; Sha, Y.; Zhang, W.; Wang, K.; Gong, J.; Tang, N.; Huang, A.; et al. Pharmacological or transcriptional inhibition of both HDAC1 and 2 leads to cell cycle blockage and apoptosis via p21Waf1/Cip1 and p19INK4d upregulation in hepatocellular carcinoma. Cell Prolif. 2018, 51, e12447. [Google Scholar] [CrossRef]
- Nettersheim, D.; Jostes, S.; Fabry, M.; Honecker, F.; Schumacher, V.; Kirfel, J.; Kristiansen, G.; Schorle, H. A signaling cascade including ARID1A, GADD45B and DUSP1 induces apoptosis and affects the cell cycle of germ cell cancers after romidepsin treatment. Oncotarget 2016, 7, 74931–74946. [Google Scholar] [CrossRef]
- Porter, N.J.; Christianson, D.W. Binding of the Microbial Cyclic Tetrapeptide Trapoxin A to the Class I Histone Deacetylase HDAC8. ACS Chem. Biol. 2017, 12, 2281–2286. [Google Scholar] [CrossRef]
- Mousazadeh, L.; Alizadeh, E.; Zarghami, N.; Hashemzadeh, S.; Aval, S.F.; Hasanifard, L.; Herizchi, R. Histone Deacetylase Inhibitor (Trapoxin A) Enhances Stemness Properties in Adipose Tissue Derived Mesenchymal Stem Cells. Drug Res. (Stuttg) 2018, 68, 450–456. [Google Scholar] [CrossRef]
- Zagni, C.; Floresta, G.; Monciino, G.; Rescifina, A. The Search for Potent, Small-Molecule HDACIs in Cancer Treatment: A Decade After Vorinostat. Med. Res. Rev. 2017, 37, 1373–1428. [Google Scholar] [CrossRef] [PubMed]
- Han, J.W.; Ahn, S.H.; Park, S.H.; Wang, S.Y.; Bae, G.U.; Seo, D.W.; Kwon, H.K.; Hong, S.; Lee, H.Y.; Lee, Y.W.; et al. Apicidin, a histone deacetylase inhibitor, inhibits proliferation of tumor cells via induction of p21WAF1/Cip1 and gelsolin. Cancer Res. 2000, 60, 6068–6074. [Google Scholar] [PubMed]
- Ahn, M.-Y. HDAC inhibitor apicidin suppresses murine oral squamous cell carcinoma cell growth in vitro and in vivo via inhibiting HDAC8 expression. Oncol. Lett. 2018, 16, 6552–6560. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Cai, D.; Zhang, B.; Lou, G.; Zou, X. Combination of HDAC inhibitor TSA and silibinin induces cell cycle arrest and apoptosis by targeting survivin and cyclinB1/Cdk1 in pancreatic cancer cells. Biomed. Pharmacother. 2015, 74, 257–264. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Q.; Liu, G.; Huang, H.; Lin, W.; Huang, Y.; Chi, Z.; Chen, S.; Lan, K.; Lin, J.; et al. Effect of histone deacetylase on prostate carcinoma. Int. J. Clin Exp. Pathol. 2015, 8, 15030–15034. [Google Scholar]
- Zhang, F.; Zhang, T.; Teng, Z.-H.; Zhang, R.; Wang, J.-B.; Mei, Q.-B. Sensitization to gamma-irradiation-induced cell cycle arrest and apoptosis by the histone deacetylase inhibitor trichostatin A in non-small cell lung cancer (NSCLC) cells. Cancer Biol. Ther. 2009, 8, 823–831. [Google Scholar] [CrossRef]
- Deng, R.; Zhang, P.; Liu, W.; Zeng, X.; Ma, X.; Shi, L.; Wang, T.; Yin, Y.; Chang, W.; Zhang, P.; et al. HDAC is indispensable for IFN-γ-induced B7-H1 expression in gastric cancer. Clin. Epigenetics 2018, 10, 153. [Google Scholar] [CrossRef]
- Di Costanzo, A.; Del Gaudio, N.; Conte, L.; Dell’Aversana, C.; Vermeulen, M.; de Thé, H.; Migliaccio, A.; Nebbioso, A.; Altucci, L. The HDAC inhibitor SAHA regulates CBX2 stability via a SUMO-triggered ubiquitin-mediated pathway in leukemia. Oncogene 2018, 37, 2559–2572. [Google Scholar] [CrossRef]
- Lee, H.-Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X.-H.; et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Horwitz, S.; Masszi, T.; Van Hoof, A.; Brown, P.; Doorduijn, J.; Hess, G.; Jurczak, W.; Knoblauch, P.; Chawla, S.; et al. Belinostat in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J. Clin. Oncol. 2015, 33, 2492–2499. [Google Scholar] [CrossRef]
- Chien, W.; Lee, D.H.; Zheng, Y.; Wuensche, P.; Alvarez, R.; Wen, D.L.; Aribi, A.M.; Thean, S.M.; Doan, N.B.; Said, J.W.; et al. Growth inhibition of pancreatic cancer cells by histone deacetylase inhibitor belinostat through suppression of multiple pathways including HIF, NFkB, and mTOR signaling in vitro and in vivo. Mol. Carcinog. 2014, 53, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Llopiz, D.; Ruiz, M.; Villanueva, L.; Iglesias, T.; Silva, L.; Egea, J.; Lasarte, J.J.; Pivette, P.; Trochon-Joseph, V.; Vasseur, B.; et al. Enhanced anti-tumor efficacy of checkpoint inhibitors in combination with the histone deacetylase inhibitor Belinostat in a murine hepatocellular carcinoma model. Cancer Immunol. Immunother. 2019, 68, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Chen, S.; Zhang, Y.; Kmieciak, M.; Leng, Y.; Li, L.; Lin, H.; Rizzo, K.A.; Dumur, C.I.; Ferreira-Gonzalez, A.; et al. The NAE inhibitor pevonedistat interacts with the HDAC inhibitor belinostat to target AML cells by disrupting the DDR. Blood 2016, 127, 2219–2230. [Google Scholar] [CrossRef]
- Cheng, Y.-W.; Liao, L.-D.; Yang, Q.; Chen, Y.; Nie, P.-J.; Zhang, X.-J.; Xie, J.-J.; Shan, B.-E.; Zhao, L.-M.; Xu, L.-Y.; et al. The histone deacetylase inhibitor panobinostat exerts anticancer effects on esophageal squamous cell carcinoma cells by inducing cell cycle arrest. Cell Biochem. Funct. 2018, 36, 398–407. [Google Scholar] [CrossRef]
- Wang, L.; Syn, N.L.-X.; Subhash, V.V.; Any, Y.; Thuya, W.L.; Cheow, E.S.H.; Kong, L.; Yu, F.; Peethala, P.C.; Wong, A.L.-A.; et al. Pan-HDAC inhibition by panobinostat mediates chemosensitization to carboplatin in non-small cell lung cancer via attenuation of EGFR signaling. Cancer Lett. 2018, 417, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Helland, Ø.; Popa, M.; Bischof, K.; Gjertsen, B.T.; McCormack, E.; Bjørge, L. The HDACi Panobinostat Shows Growth Inhibition Both In Vitro and in a Bioluminescent Orthotopic Surgical Xenograft Model of Ovarian Cancer. PLoS ONE 2016, 11, e0158208. [Google Scholar] [CrossRef] [PubMed]
- Hennika, T.; Hu, G.; Olaciregui, N.G.; Barton, K.L.; Ehteda, A.; Chitranjan, A.; Chang, C.; Gifford, A.J.; Tsoli, M.; Ziegler, D.S.; et al. Pre-Clinical Study of Panobinostat in Xenograft and Genetically Engineered Murine Diffuse Intrinsic Pontine Glioma Models. PLoS ONE 2017, 12, e0169485. [Google Scholar] [CrossRef] [PubMed]
- Topper, M.J.; Vaz, M.; Chiappinelli, K.B.; DeStefano Shields, C.E.; Niknafs, N.; Yen, R.-W.C.; Wenzel, A.; Hicks, J.; Ballew, M.; Stone, M.; et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell 2017, 171, 1284–1300. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, K.; Yao, C.; Kahwash, S.; Tang, Y.; Zhang, G.; Patterson, K.; Wang, Q.-E.; Zhao, W. Givinostat, a type II histone deacetylase inhibitor, induces potent caspase-dependent apoptosis in human lymphoblastic leukemia. Genes Cancer 2016, 7, 292–300. [Google Scholar] [CrossRef]
- Angeletti, F.; Fossati, G.; Pattarozzi, A.; Würth, R.; Solari, A.; Daga, A.; Masiello, I.; Barbieri, F.; Florio, T.; Comincini, S. Inhibition of the Autophagy Pathway Synergistically Potentiates the Cytotoxic Activity of Givinostat (ITF2357) on Human Glioblastoma Cancer Stem Cells. Front. Mol. Neurosci. 2016, 9, 107. [Google Scholar] [CrossRef]
- Pinazza, M.; Borga, C.; Agnusdei, V.; Minuzzo, S.; Fossati, G.; Paganin, M.; Michielotto, B.; De Paoli, A.; Basso, G.; Amadori, A.; et al. An immediate transcriptional signature associated with response to the histone deacetylase inhibitor Givinostat in T acute lymphoblastic leukemia xenografts. Cell Death Dis. 2016, 6, e2047. [Google Scholar] [CrossRef] [PubMed]
- Muscolini, M.; Castiello, L.; Palermo, E.; Zevini, A.; Ferrari, M.; Olagnier, D.; Hiscott, J. SIRT1 Modulates the Sensitivity of Prostate Cancer Cells to Vesicular Stomatitis Virus Oncolysis. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Ohno, I.; Ueno, H.; Mitsunaga, S.; Hashimoto, Y.; Okusaka, T.; Kondo, S.; Sasaki, M.; Sakamoto, Y.; Takahashi, H.; et al. Phase I study of resminostat, an HDAC inhibitor, combined with S-1 in patients with pre-treated biliary tract or pancreatic cancer. Invest. New Drugs 2019, 37, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Tambo, Y.; Hosomi, Y.; Sakai, H.; Nogami, N.; Atagi, S.; Sasaki, Y.; Kato, T.; Takahashi, T.; Seto, T.; Maemondo, M.; et al. Phase I/II study of docetaxel combined with resminostat, an oral hydroxamic acid HDAC inhibitor, for advanced non-small cell lung cancer in patients previously treated with platinum-based chemotherapy. Invest. New Drugs 2017, 35, 217–226. [Google Scholar] [CrossRef]
- Bitzer, M.; Horger, M.; Giannini, E.G.; Ganten, T.M.; Wörns, M.A.; Siveke, J.T.; Dollinger, M.M.; Gerken, G.; Scheulen, M.E.; Wege, H.; et al. Resminostat plus sorafenib as second-line therapy of advanced hepatocellular carcinoma - The SHELTER study. J. Hepatol. 2016, 65, 280–288. [Google Scholar] [CrossRef]
- Salvador, M.A.; Wicinski, J.; Cabaud, O.; Toiron, Y.; Finetti, P.; Josselin, E.; Lelièvre, H.; Kraus-Berthier, L.; Depil, S.; Bertucci, F.; et al. The histone deacetylase inhibitor abexinostat induces cancer stem cells differentiation in breast cancer with low Xist expression. Clin. Cancer Res. 2013, 19, 6520–6531. [Google Scholar] [CrossRef]
- Sholler, G.S.; Currier, E.A.; Dutta, A.; Slavik, M.A.; Illenye, S.A.; Mendonca, M.C.F.; Dragon, J.; Roberts, S.S.; Bond, J.P. PCI-24781 (abexinostat), a novel histone deacetylase inhibitor, induces reactive oxygen species-dependent apoptosis and is synergistic with bortezomib in neuroblastoma. J. Cancer Ther. Res. 2013, 2, 21. [Google Scholar] [CrossRef]
- Bhalla, S.; Evens, A.M.; Prachand, S.; Schumacker, P.T.; Gordon, L.I. Paradoxical regulation of hypoxia inducible factor-1α (HIF-1α) by histone deacetylase inhibitor in diffuse large B-cell lymphoma. PLoS ONE 2013, 8, e81333. [Google Scholar] [CrossRef]
- Fukuda, K.; Takeuchi, S.; Arai, S.; Katayama, R.; Nanjo, S.; Tanimoto, A.; Nishiyama, A.; Nakagawa, T.; Taniguchi, H.; Suzuki, T.; et al. Epithelial-to-Mesenchymal Transition Is a Mechanism of ALK Inhibitor Resistance in Lung Cancer Independent of ALK Mutation Status. Cancer Res. 2019, 79, 1658–1670. [Google Scholar] [CrossRef]
- Stühmer, T.; Arts, J.; Chatterjee, M.; Borawski, J.; Wolff, A.; King, P.; Einsele, H.; Leo, E.; Bargou, R.C. Preclinical anti-myeloma activity of the novel HDAC-inhibitor JNJ-26481585. Br. J. Haematol. 2010, 149, 529–536. [Google Scholar] [CrossRef]
- Haydn, T.; Metzger, E.; Schuele, R.; Fulda, S. Concomitant epigenetic targeting of LSD1 and HDAC synergistically induces mitochondrial apoptosis in rhabdomyosarcoma cells. Cell Death Dis. 2017, 8, e2879. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Abaza, Y.; Takahashi, K.; Medeiros, B.C.; Arellano, M.; Khaled, S.K.; Patnaik, M.; Odenike, O.; Sayar, H.; Tummala, M.; et al. Pracinostat plus azacitidine in older patients with newly diagnosed acute myeloid leukemia: Results of a phase 2 study. Blood Adv. 2019, 3, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Montalban-Bravo, G.; Berdeja, J.G.; Abaza, Y.; Jabbour, E.; Essell, J.; Lyons, R.M.; Ravandi, F.; Maris, M.; Heller, B.; et al. Phase 2, randomized, double-blind study of pracinostat in combination with azacitidine in patients with untreated, higher-risk myelodysplastic syndromes. Cancer 2017, 123, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Swaminathan, M.; Pemmaraju, N.; Ferrajoli, A.; Jabbour, E.J.; Daver, N.G.; DiNardo, C.D.; Alvarado, Y.; Yilmaz, M.; Huynh-Lu, J.; et al. A phase 2 study of pracinostat combined with ruxolitinib in patients with myelofibrosis. Leuk. Lymphoma 2019, 60, 1767–1774. [Google Scholar] [CrossRef]
- Amengual, J.E.; Prabhu, S.A.; Lombardo, M.; Zullo, K.; Johannet, P.M.; Gonzalez, Y.; Scotto, L.; Serrano, X.J.; Wei, Y.; Duong, J.; et al. Mechanisms of Acquired Drug Resistance to the HDAC6 Selective Inhibitor Ricolinostat Reveals Rational Drug-Drug Combination with Ibrutinib. Clin. Cancer Res. 2017, 23, 3084–3096. [Google Scholar] [CrossRef]
- Cao, J.; Lv, W.; Wang, L.; Xu, J.; Yuan, P.; Huang, S.; He, Z.; Hu, J. Ricolinostat (ACY-1215) suppresses proliferation and promotes apoptosis in esophageal squamous cell carcinoma via miR-30d/PI3K/AKT/mTOR and ERK pathways. Cell Death Dis. 2018, 9, 817. [Google Scholar] [CrossRef]
- Adeegbe, D.O.; Liu, Y.; Lizotte, P.H.; Kamihara, Y.; Aref, A.R.; Almonte, C.; Dries, R.; Li, Y.; Liu, S.; Wang, X.; et al. Synergistic Immunostimulatory Effects and Therapeutic Benefit of Combined Histone Deacetylase and Bromodomain Inhibition in Non-Small Cell Lung Cancer. Cancer Discov. 2017, 7, 852–867. [Google Scholar] [CrossRef]
- Banerji, U.; van Doorn, L.; Papadatos-Pastos, D.; Kristeleit, R.; Debnam, P.; Tall, M.; Stewart, A.; Raynaud, F.; Garrett, M.D.; Toal, M.; et al. A phase I pharmacokinetic and pharmacodynamic study of CHR-3996, an oral class I selective histone deacetylase inhibitor in refractory solid tumors. Clin. Cancer Res. 2012, 18, 2687–2694. [Google Scholar] [CrossRef]
- Smith, E.M.; Zhang, L.; Walker, B.A.; Davenport, E.L.; Aronson, L.I.; Krige, D.; Hooftman, L.; Drummond, A.H.; Morgan, G.J.; Davies, F.E. The combination of HDAC and aminopeptidase inhibitors is highly synergistic in myeloma and leads to disruption of the NFκB signalling pathway. Oncotarget 2015, 6, 17314–17327. [Google Scholar] [CrossRef]
- Li, Q.; Ding, C.; Meng, T.; Lu, W.; Liu, W.; Hao, H.; Cao, L. Butyrate suppresses motility of colorectal cancer cells via deactivating Akt/ERK signaling in histone deacetylase dependent manner. J. Pharmacol. Sci. 2017, 135, 148–155. [Google Scholar] [CrossRef]
- Su, J.M.; Li, X.-N.; Thompson, P.; Ou, C.-N.; Ingle, A.M.; Russell, H.; Lau, C.C.; Adamson, P.C.; Blaney, S.M. Phase 1 study of valproic acid in pediatric patients with refractory solid or CNS tumors: A children’s oncology group report. Clin. Cancer Res. 2011, 17, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Wheler, J.J.; Janku, F.; Falchook, G.S.; Jackson, T.L.; Fu, S.; Naing, A.; Tsimberidou, A.M.; Moulder, S.L.; Hong, D.S.; Yang, H.; et al. Phase I study of anti-VEGF monoclonal antibody bevacizumab and histone deacetylase inhibitor valproic acid in patients with advanced cancers. Cancer Chemother. Pharmacol. 2014, 73, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.F.; Karpenko, M.J.; Liu, Z.; Aimiuwu, J.; Villalona-Calero, M.A.; Chan, K.K.; Grever, M.R.; Otterson, G.A. Phase I study of 5-aza-2’-deoxycytidine in combination with valproic acid in non-small-cell lung cancer. Cancer Chemother. Pharmacol. 2013, 71, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Iwahashi, S.; Utsunomiya, T.; Imura, S.; Morine, Y.; Ikemoto, T.; Arakawa, Y.; Saito, Y.; Ishikawa, D.; Shimada, M. Effects of valproic acid in combination with S-1 on advanced pancreatobiliary tract cancers: Clinical study phases I/II. Anticancer Res. 2014, 34, 5187–5191. [Google Scholar]
- Chan, E.; Chiorean, E.G.; O’Dwyer, P.J.; Gabrail, N.Y.; Alcindor, T.; Potvin, D.; Chao, R.; Hurwitz, H. Phase I/II study of mocetinostat in combination with gemcitabine for patients with advanced pancreatic cancer and other advanced solid tumors. Cancer Chemother. Pharmacol. 2018, 81, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Oki, Y.; Bociek, R.G.; Kuruvilla, J.; Fanale, M.; Neelapu, S.; Copeland, A.; Buglio, D.; Galal, A.; Besterman, J.; et al. Mocetinostat for relapsed classical Hodgkin’s lymphoma: An open-label, single-arm, phase 2 trial. Lancet Oncol. 2011, 12, 1222–1228. [Google Scholar] [CrossRef]
- Lopez, G.; Braggio, D.; Zewdu, A.; Casadei, L.; Batte, K.; Bid, H.K.; Koller, D.; Yu, P.; Iwenofu, O.H.; Strohecker, A.; et al. Mocetinostat combined with gemcitabine for the treatment of leiomyosarcoma: Preclinical correlates. PLoS ONE 2017, 12, e0188859. [Google Scholar] [CrossRef] [PubMed]
- Karandish, F.; Haldar, M.K.; You, S.; Brooks, A.E.; Brooks, B.D.; Guo, B.; Choi, Y.; Mallik, S. Prostate-Specific Membrane Antigen Targeted Polymersomes for Delivering Mocetinostat and Docetaxel to Prostate Cancer Cell Spheroids. ACS Omega 2016, 1, 952–962. [Google Scholar] [CrossRef]
- Was, H.; Krol, S.K.; Rotili, D.; Mai, A.; Wojtas, B.; Kaminska, B.; Maleszewska, M. Histone deacetylase inhibitors exert anti-tumor effects on human adherent and stem-like glioma cells. Clin. Epigenetics 2019, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Chung, V.Y.; Tan, T.Z.; Ye, J.; Huang, R.-L.; Lai, H.-C.; Kappei, D.; Wollmann, H.; Guccione, E.; Huang, R.Y.-J. The role of GRHL2 and epigenetic remodeling in epithelial-mesenchymal plasticity in ovarian cancer cells. Commun. Biol. 2019, 2, 272. [Google Scholar] [CrossRef] [PubMed]
- Yeruva, S.L.H.; Zhao, F.; Miller, K.D.; Tevaarwerk, A.J.; Wagner, L.I.; Gray, R.J.; Sparano, J.A.; Connolly, R.M. E2112: Randomized phase iii trial of endocrine therapy plus entinostat/placebo in patients with hormone receptor-positive advanced breast cancer. NPJ Breast Cancer 2018, 4, 1. [Google Scholar] [CrossRef]
- Suzuki, T.; Ando, T.; Tsuchiya, K.; Fukazawa, N.; Saito, A.; Mariko, Y.; Yamashita, T.; Nakanishi, O. Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives. J. Med. Chem. 1999, 42, 3001–3003. [Google Scholar] [CrossRef]
- Ahn, M.Y.; Kang, D.O.; Na, Y.J.; Yoon, S.; Choi, W.S.; Kang, K.W.; Chung, H.Y.; Jung, J.H.; Min, D.S.; Kim, H.S. Histone deacetylase inhibitor, apicidin, inhibits human ovarian cancer cell migration via class II histone deacetylase 4 silencing. Cancer Lett. 2012, 325, 189–199. [Google Scholar] [CrossRef]
- Ansari, D.; Urey, C.; Hilmersson, K.S.; Bauden, M.P.; Ek, F.; Olsson, R.; Andersson, R. Apicidin sensitizes pancreatic cancer cells to gemcitabine by epigenetically regulating MUC4 expression. Anticancer Res. 2014, 34, 5269–5276. [Google Scholar]
- Salvador, L.A.; Park, H.; Al-Awadhi, F.H.; Liu, Y.; Kim, B.; Zeller, S.L.; Chen, Q.-Y.; Hong, J.; Luesch, H. Modulation of Activity Profiles for Largazole-Based HDAC Inhibitors through Alteration of Prodrug Properties. ACS Med. Chem. Lett. 2014, 5, 905–910. [Google Scholar] [CrossRef] [PubMed]
- Pilon, J.L.; Clausen, D.J.; Hansen, R.J.; Lunghofer, P.J.; Charles, B.; Rose, B.J.; Thamm, D.H.; Gustafson, D.L.; Bradner, J.E.; Williams, R.M. Comparative pharmacokinetic properties and antitumor activity of the marine HDACi Largazole and Largazole peptide isostere. Cancer Chemother. Pharmacol. 2015, 75, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.-C.; Wen, Z.-S.; Qiu, Y.-T.; Chen, X.-Q.; Chen, H.-B.; Wei, M.-M.; Liu, Z.; Jiang, S.; Zhou, G.-B. Largazole Arrests Cell Cycle at G1 Phase and Triggers Proteasomal Degradation of E2F1 in Lung Cancer Cells. ACS Med. Chem. Lett 2013, 4, 921–926. [Google Scholar] [CrossRef]
- Takamura, T.; Horinaka, M.; Yasuda, S.; Toriyama, S.; Aono, Y.; Sowa, Y.; Miki, T.; Ukimura, O.; Sakai, T. FGFR inhibitor BGJ398 and HDAC inhibitor OBP-801 synergistically inhibit cell growth and induce apoptosis in bladder cancer cells. Oncol. Rep. 2018, 39, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Crabb, S.J.; Howell, M.; Rogers, H.; Ishfaq, M.; Yurek-George, A.; Carey, K.; Pickering, B.M.; East, P.; Mitter, R.; Maeda, S.; et al. Characterisation of the in vitro activity of the depsipeptide histone deacetylase inhibitor spiruchostatin A. Biochem. Pharmacol. 2008, 76, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Maulucci, N.; Chini, M.G.; Micco, S.D.; Izzo, I.; Cafaro, E.; Russo, A.; Gallinari, P.; Paolini, C.; Nardi, M.C.; Casapullo, A.; et al. Molecular insights into azumamide e histone deacetylases inhibitory activity. J. Am. Chem. Soc. 2007, 129, 3007–3012. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, X.; He, K.; Xiao, J.; Duan, X.; Huang, R.; Xia, Z.; He, J.; Zhang, J.; Xiang, G. Histone deacetylase inhibitor screening identifies HC toxin as the most effective in intrahepatic cholangiocarcinoma cells. Oncol. Rep. 2016, 35, 2535–2542. [Google Scholar] [CrossRef] [PubMed]
- Min, K.N.; Cho, M.J.; Kim, D.-K.; Sheen, Y.Y. Estrogen receptor enhances the antiproliferative effects of trichostatin A and HC-toxin in human breast cancer cells. Arch. Pharm. Res. 2004, 27, 554–561. [Google Scholar] [CrossRef]
- Deubzer, H.E.; Ehemann, V.; Westermann, F.; Heinrich, R.; Mechtersheimer, G.; Kulozik, A.E.; Schwab, M.; Witt, O. Histone deacetylase inhibitor Helminthosporium carbonum (HC)-toxin suppresses the malignant phenotype of neuroblastoma cells. Int. J. Cancer 2008, 122, 1891–1900. [Google Scholar] [CrossRef]
- Milde, T.; Lodrini, M.; Savelyeva, L.; Korshunov, A.; Kool, M.; Brueckner, L.M.; Antunes, A.S.L.M.; Oehme, I.; Pekrun, A.; Pfister, S.M.; et al. HD-MB03 is a novel Group 3 medulloblastoma model demonstrating sensitivity to histone deacetylase inhibitor treatment. J. Neurooncol. 2012, 110, 335–348. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Csizmadia, P. MarvinSketch and MarvinView: Molecule Applets for the World Wide Web. In Proceedings of the 3rd International Electronic Conference on Synthetic Organic Chemistry, Budapest, Hungary, 1–30 September 1999; MDPI: Basel, Switzerland, 1999; p. 1775. [Google Scholar]
- Shigematsu, H. Electron cryo-microscopy for elucidating the dynamic nature of live-protein complexes. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129436. [Google Scholar] [CrossRef] [PubMed]
- Kalin, J.H.; Wu, M.; Gomez, A.V.; Song, Y.; Das, J.; Hayward, D.; Adejola, N.; Wu, M.; Panova, I.; Chung, H.J.; et al. Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nat. Commun. 2018, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Karoutas, A.; Szymanski, W.; Rausch, T.; Guhathakurta, S.; Rog-Zielinska, E.A.; Peyronnet, R.; Seyfferth, J.; Chen, H.-R.; de Leeuw, R.; Herquel, B.; et al. The NSL complex maintains nuclear architecture stability via lamin A/C acetylation. Nat. Cell Biol. 2019, 21, 1248–1260. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Ye, S.; Li, K.; Huang, M.; Wang, Q.; Zeng, S.; Chen, X.; Gao, W.; Chen, J.; Zhang, Q.; et al. NOS1 inhibits the interferon response of cancer cells by S-nitrosylation of HDAC2. J. Exp. Clin. Cancer Res. 2019, 38, 483. [Google Scholar] [CrossRef]







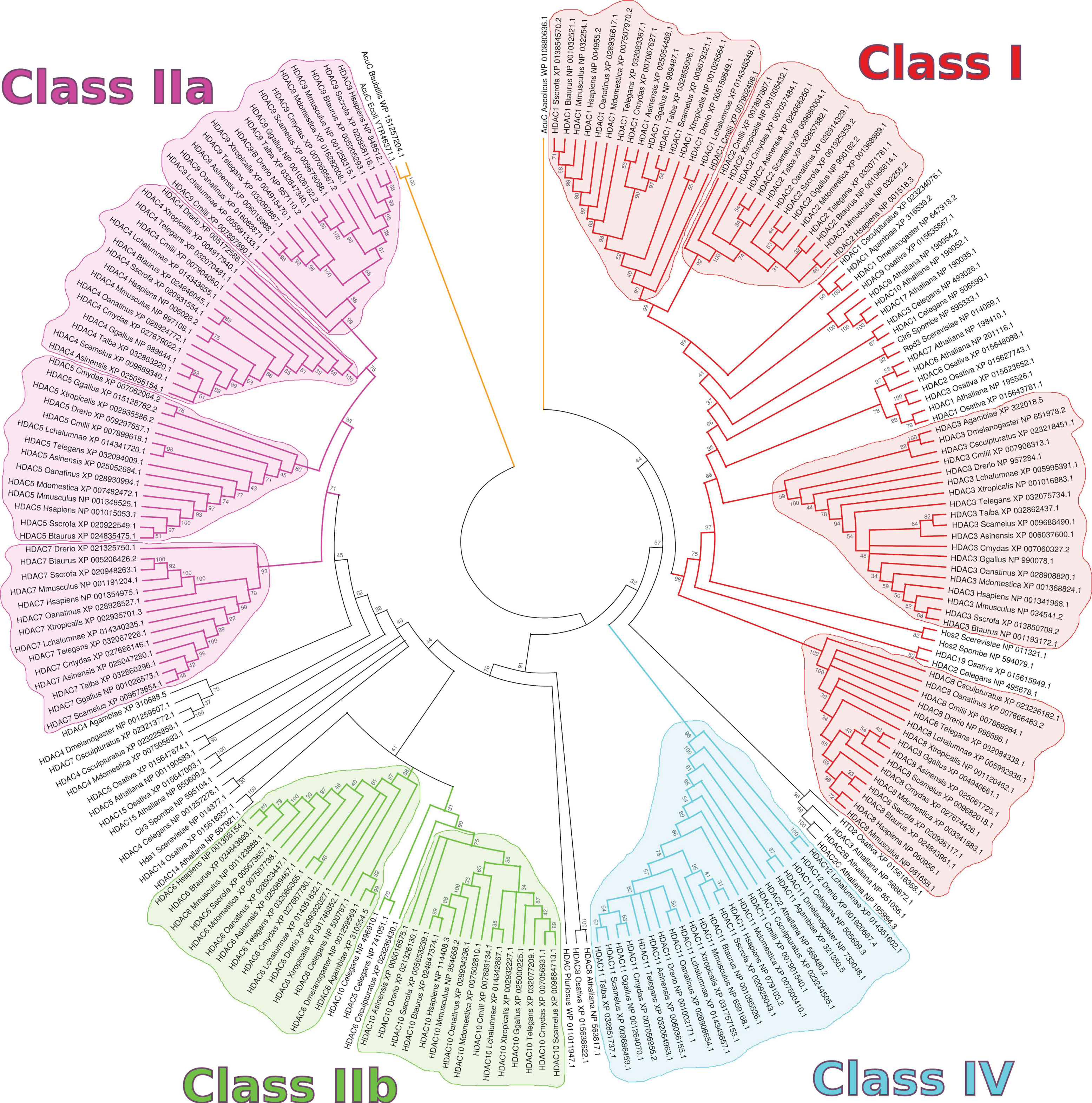{kind=link}
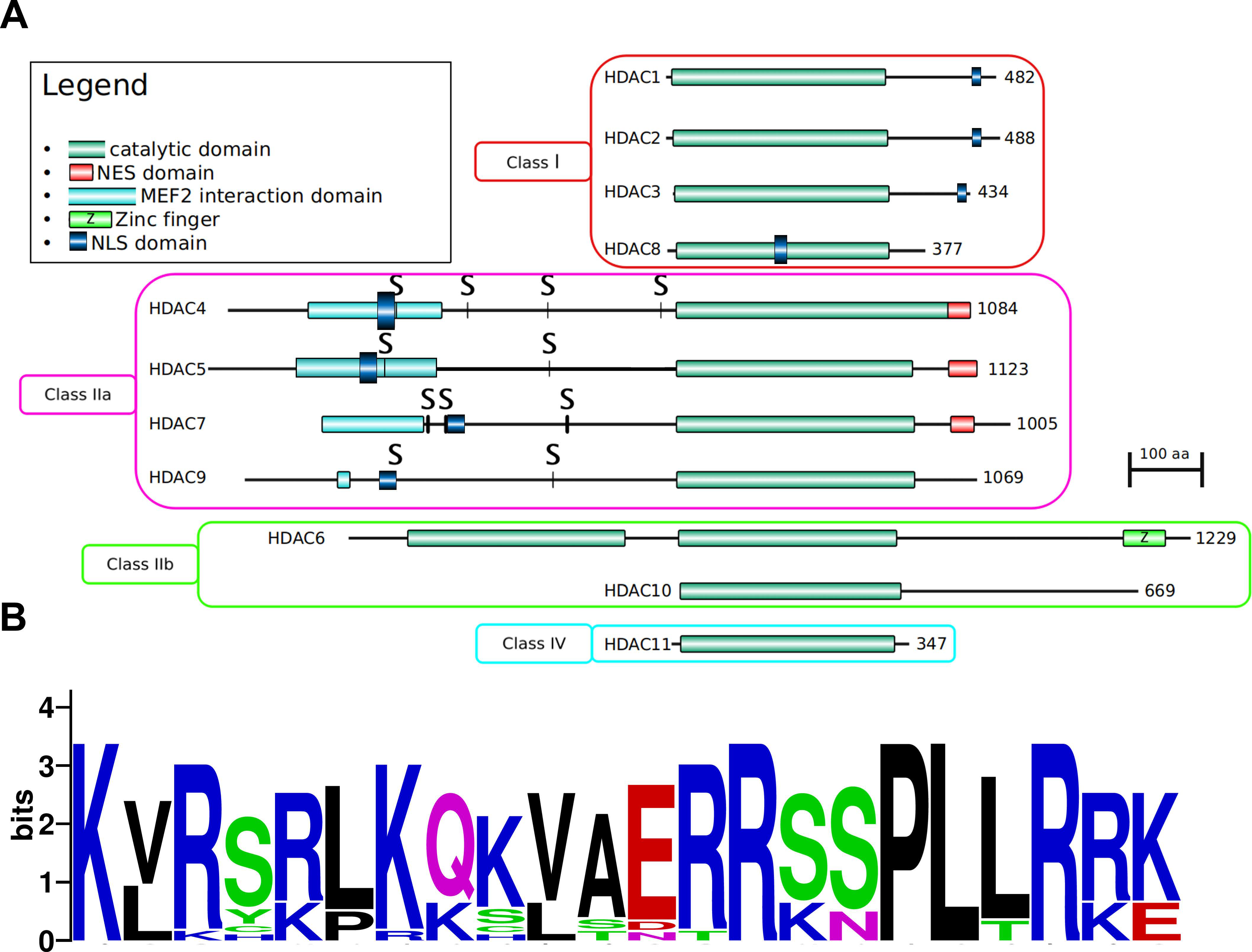{kind=link}
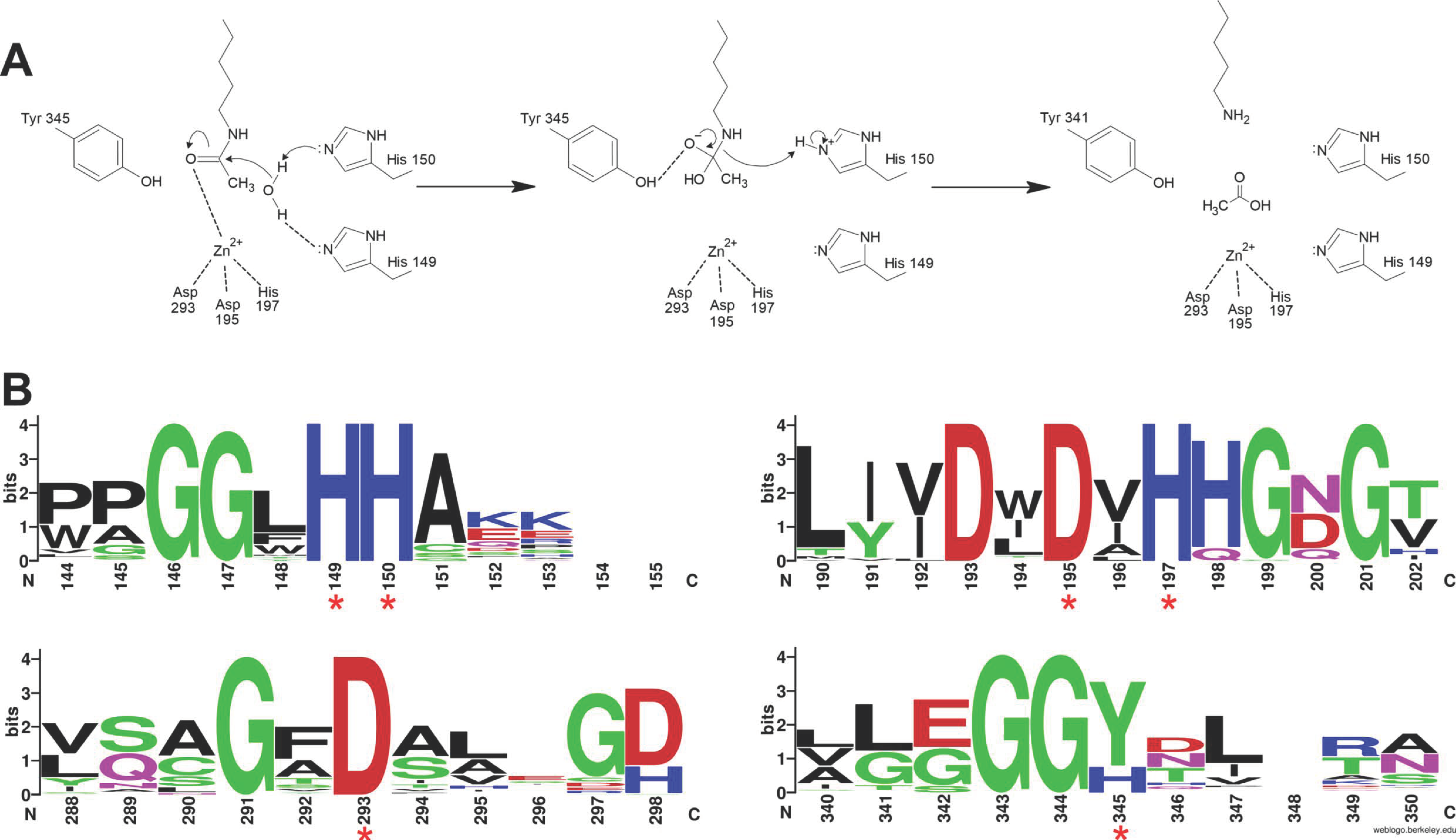{kind=link}
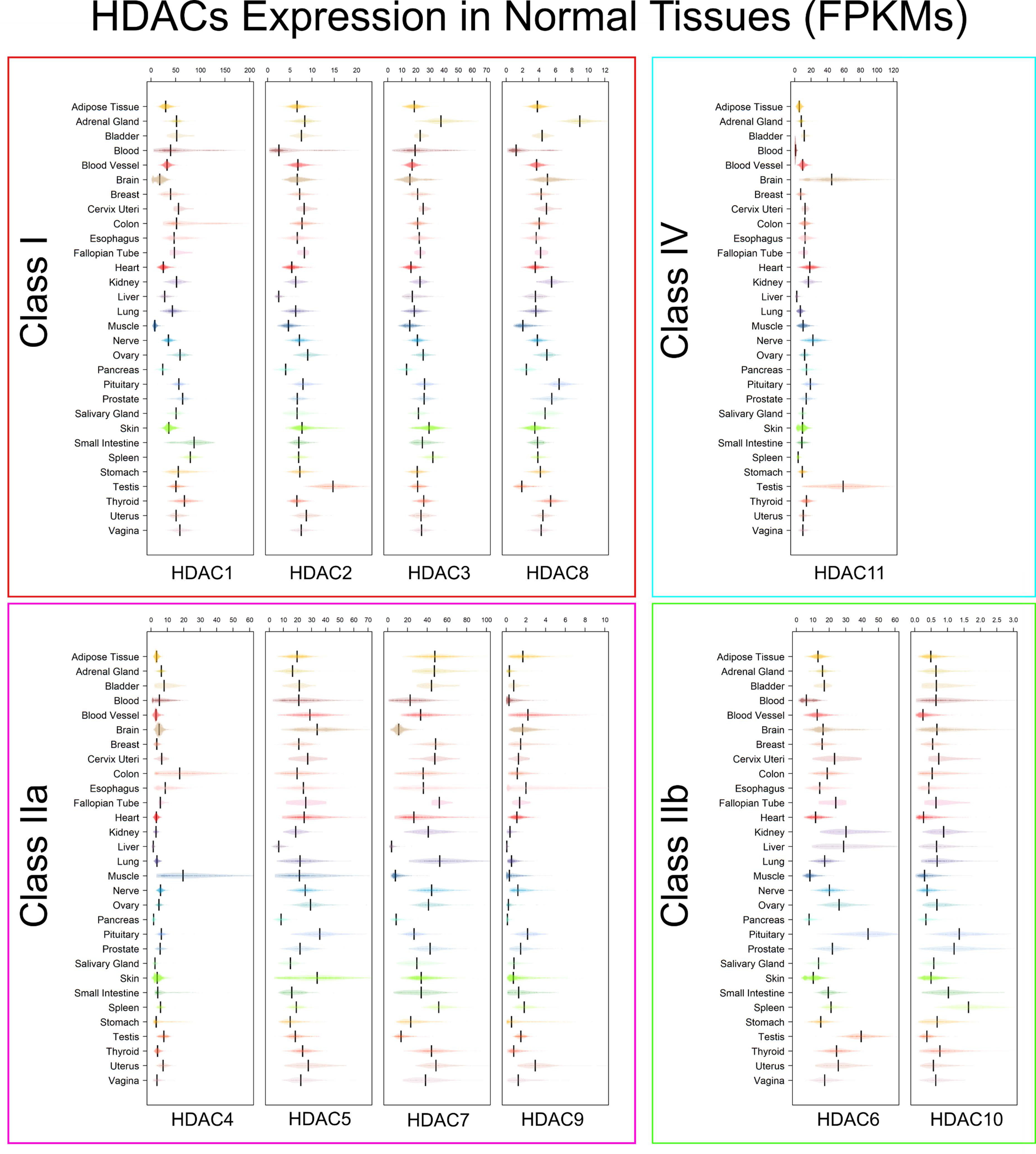{kind=link}
{kind=link}
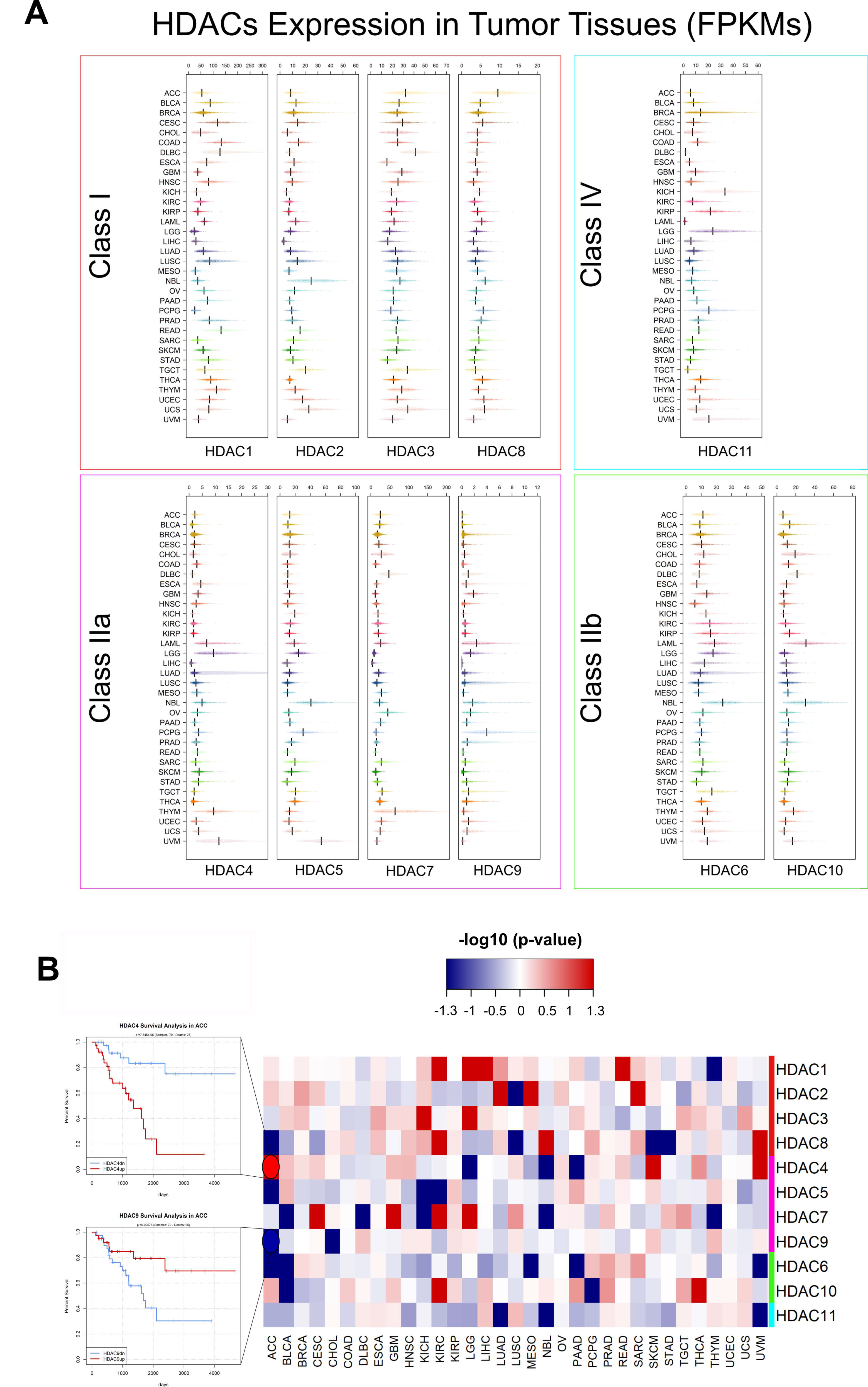{kind=link}
| Family | Class (Yeast Homolog) | Subclass | Protein | Cell Compartment |
|---|---|---|---|---|
| Classical (Zn2+ dependent) | I (Rpd3) | HDAC1 | Nucleus | |
| HDAC2 | Nucleus | |||
| HDAC3 | Nucleus | |||
| HDAC8 | Nucleus | |||
| II (Hda1) | IIa | HDAC4 | Cytoplasm/Nucleus | |
| HDAC5 | Cytoplasm/Nucleus | |||
| HDAC7 | Cytoplasm/Nucleus | |||
| HDAC9 | Cytoplasm/Nucleus | |||
| IIb | HDAC6 | Cytoplasm | ||
| HDAC10 | Cytoplasm | |||
| IV (Rpd3, Hda1) | HDAC11 | Cytoplasm/Nucleus | ||
| NAD dependent | III (Sir2, Hst1-4) | SIRT 1-7 | Cytoplasm/Nucleus |
| Organism | Nr. of HDACs |
|---|---|
| Homo sapiens (human) | 11 |
| Mus musculus (mouse) | 11 |
| Bos taurus (cattle) | 11 |
| Sus scrofa (pig) | 11 |
| Monodelphis domestica (opossum) | 10 |
| Ornithorhyncus anatinus (platypus) | 11 |
| Gallus gallus (chicken) | 10 |
| Tyto alba (barn owl) | 7 |
| Struthio camelus (ostrich) | 9 |
| Alligator sinensis (alligator) | 11 |
| Thamnophis elegans (snake) | 11 |
| Chelonia mydas (turtle) | 11 |
| Xenopus tropicalis (frog) | 11 |
| Danio rerio (zebrafish) | 11 |
| Latimeria chalumnae (coealacanth) | 11 |
| Callorhinchus milii (shark) | 9 |
| Anopheles gambiae (mosquito) | 5 |
| Drosophila melanogaster (fruit fly) | 5 |
| Centruroides sculpturatus (scorpion) | 7 |
| Caenorhabditis elegans (nematode) | 8 |
| Arabidopsis thaliana (thale cress) | 14 |
| Oryza sativa (rice) | 11 |
| Schizosaccharomices pombe (fission yeast) | 3 |
| Saccharomyces cerevisiae (budding yeast) | 3 |
| Pyrococcus furiosus (archaeon) | 1 |
| Structural Group | Name | Specificity | Cancer | Reference |
|---|---|---|---|---|
| Group I - Hydroxamic acids | ||||
 | Trichostatin A (TSA) | Pan | Pancreatic, prostate, NSCLC, gastric | [265,266,267,268] |
 | Vorinostat (SAHA) | Pan | Ovarian, gastric, leukemia | [221,268,269] |
 | Belinostat (PXD101) | Pan | T-Cell lymphoma, pancreatic, hepatocellular carcinoma, acute myelogenous leukemia | [270,271,272,273,274] |
 | Panobinostat (LBH589) | Pan | MLL-ALL, neuroblastoma, esophageal squamous cell carcinoma, NSCLC, ovarian, pontine glioma, DLBCL | [216,223,227,275,276,277,278] |
 | Givinostat (ITF-2357) | Pan | NSCLC, acute lymphoblastic leukemia, glioblastoma, acute lymphoblastic leukemia | [279,280,281,282] |
 | Resminostat (RAS2410) | Pan | Prostate, pancreas, NSCLC, hepatocellular carcinoma | [283,284,285,286] |
 | Abexinostat (PCI-24781) | Pan | breast, gastric, neuroblastoma, DLBCL | [267,287,288,289] |
 | Quisinostat (JNJ-26481585) | Pan | Lung, multiple myeloma, rhabdomyosarcoma | [290,291,292] |
 | Pracinostat (SB939) | Pan | Acute lymphoblastic leukemia, myelodysplastic syndromes, myelofibrosis, NSCLC | [222,293,294,295] |
 | Rocilinostat (ACY-1215) | HDAC6 | Esophageal squamous cell carcinoma, DLBCL, multiple myeloma, NSCLC | [229,296,297,298] |
 | Nanatinostat (CHR-3996) | Class I & II | HCC, multiple myeloma | [228,299,300] |
| Group II—SCFAs | ||||
 | Sodium butyrate and derivatives | Class I & IIa | Breast, colorectal, gastric | [235,268,301] |
 | Valproic acid (VPA) | Class I | CNS, colorectal, prostate cancer, NSCLC, pancreatobiliary tract | [302,303,304,305] |
| Group III—Benzamides | ||||
 | Mocetinostat (MGCD0103) | Class I & IV | Pancreatic, Hodgkin’s lymphoma, SHH medulloblastoma, leiomyosarcoma, prostate, glioblastoma, ovarian | [197,306,307,308,309,310,311] |
 | Entinostat (MS-275) | Class I & IV | Breast, leukemia, ovarian, renal cell carcinoma | [244,245,312,313] |
 | K560 and derivatives | Class I | N/A | [248,249] |
| Group IV—Tetrapeptides (depsipeptides) | ||||
 | Apicidin (OSI-2040) | Class I | Oral squamous cell carcinoma, ovarian, pancreatic | [264,314,315] |
 | Romidepsin (FK228) | Class I | Germ cell, HCC, T cell lymphoma | [256,257,259] |
 | Chromopeptide A | Class I | Prostate | [255] |
 | Largazole | Class I | Colon, lung | [316,317,318] |
 | Spiruchostatin A | Class I | Bladder, breast | [319,320] |
| Tetrapeptides (α4-cyclotetrapeptides) | ||||
 | Trapoxin | HDAC8 | N/A | [252,261] |
 | Azumamide A and derivates | Class I | N/A | [321] |
 | HC-toxin | Class I | Cholangiocarcinoma, breast, neuroblastoma, medulloblastoma | [322,323,324,325] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milazzo, G.; Mercatelli, D.; Di Muzio, G.; Triboli, L.; De Rosa, P.; Perini, G.; Giorgi, F.M. Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes 2020, 11, 556. https://doi.org/10.3390/genes11050556
Milazzo G, Mercatelli D, Di Muzio G, Triboli L, De Rosa P, Perini G, Giorgi FM. Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes. 2020; 11(5):556. https://doi.org/10.3390/genes11050556
Chicago/Turabian StyleMilazzo, Giorgio, Daniele Mercatelli, Giulia Di Muzio, Luca Triboli, Piergiuseppe De Rosa, Giovanni Perini, and Federico M. Giorgi. 2020. "Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability" Genes 11, no. 5: 556. https://doi.org/10.3390/genes11050556
APA StyleMilazzo, G., Mercatelli, D., Di Muzio, G., Triboli, L., De Rosa, P., Perini, G., & Giorgi, F. M. (2020). Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes, 11(5), 556. https://doi.org/10.3390/genes11050556