Pitfalls in Single Clone CRISPR-Cas9 Mutagenesis to Fine-Map Regulatory Intervals


 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. eGenes, Candidate eSNPs, and Control SNP Selection
2.2. SNP-Targeting and gRNA Screening Design
2.3. Single-Cell Clone Generation
2.4. Myeloid Lineage Differentiation
2.5. Flow Cytometry
2.6. Immunofluorescence
2.7. RNA Isolation
2.8. Bulk RNA-Seq and Differential Gene Expression Analysis
2.9. Variant Calling
2.10. Fluidigm qRT-PCR
2.11. Plasmid Construction
2.12. CRISPR-Edited Single-Cell Clones Generation
2.13. Power Simulation Studies
3. Results
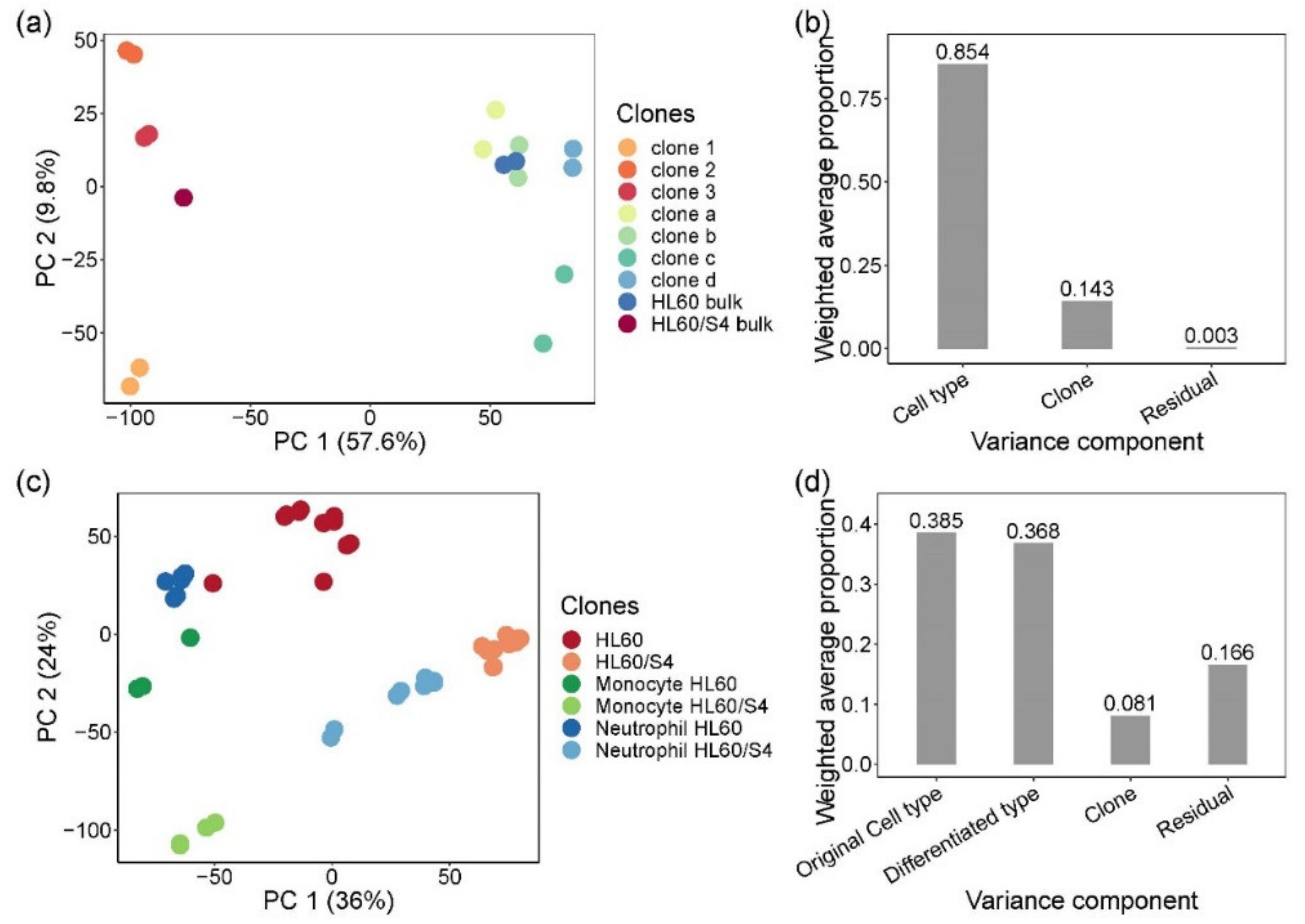
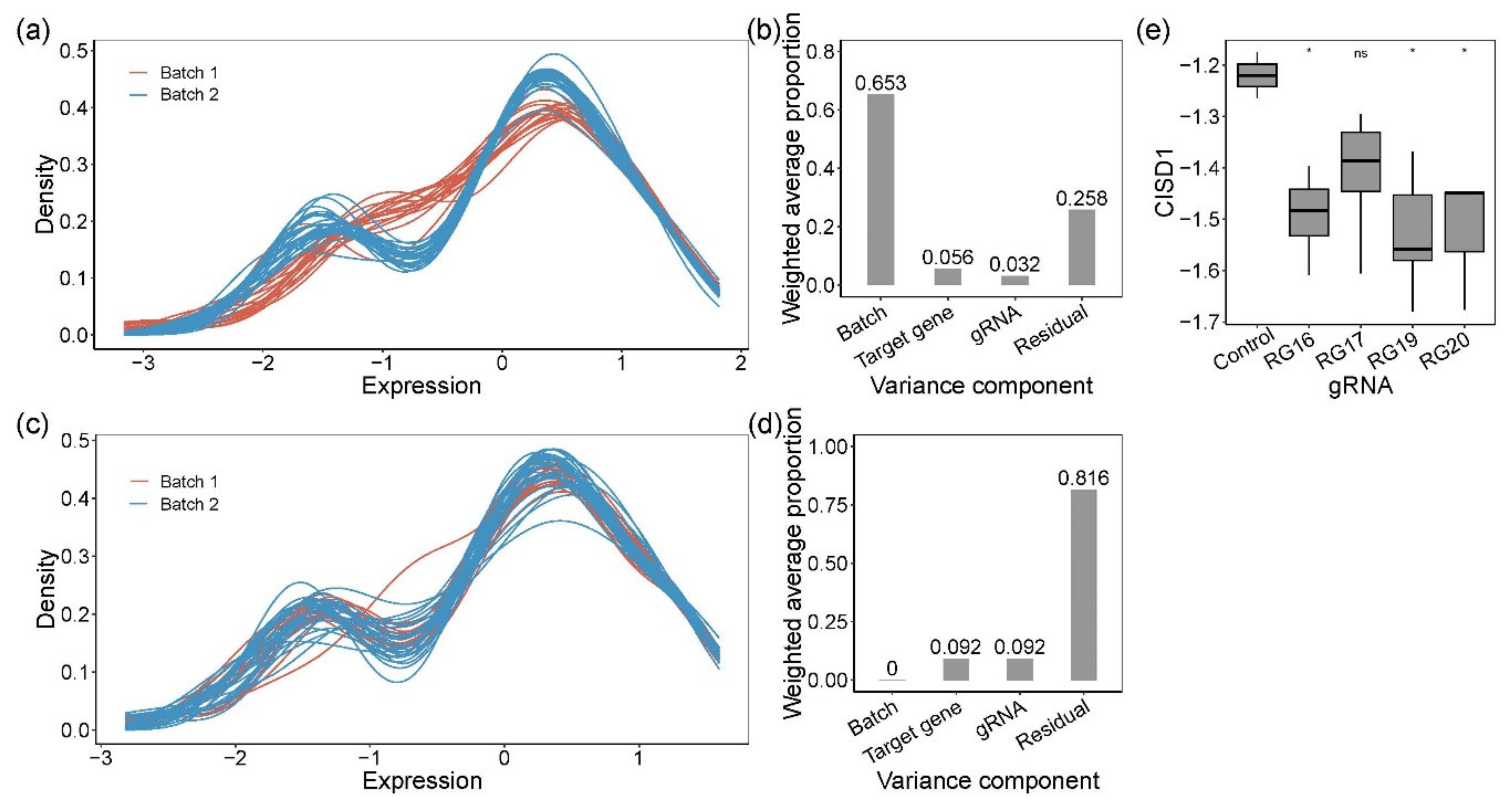
3.1. Effect of Clonal Variability on Gene Expression in HL60 Cells
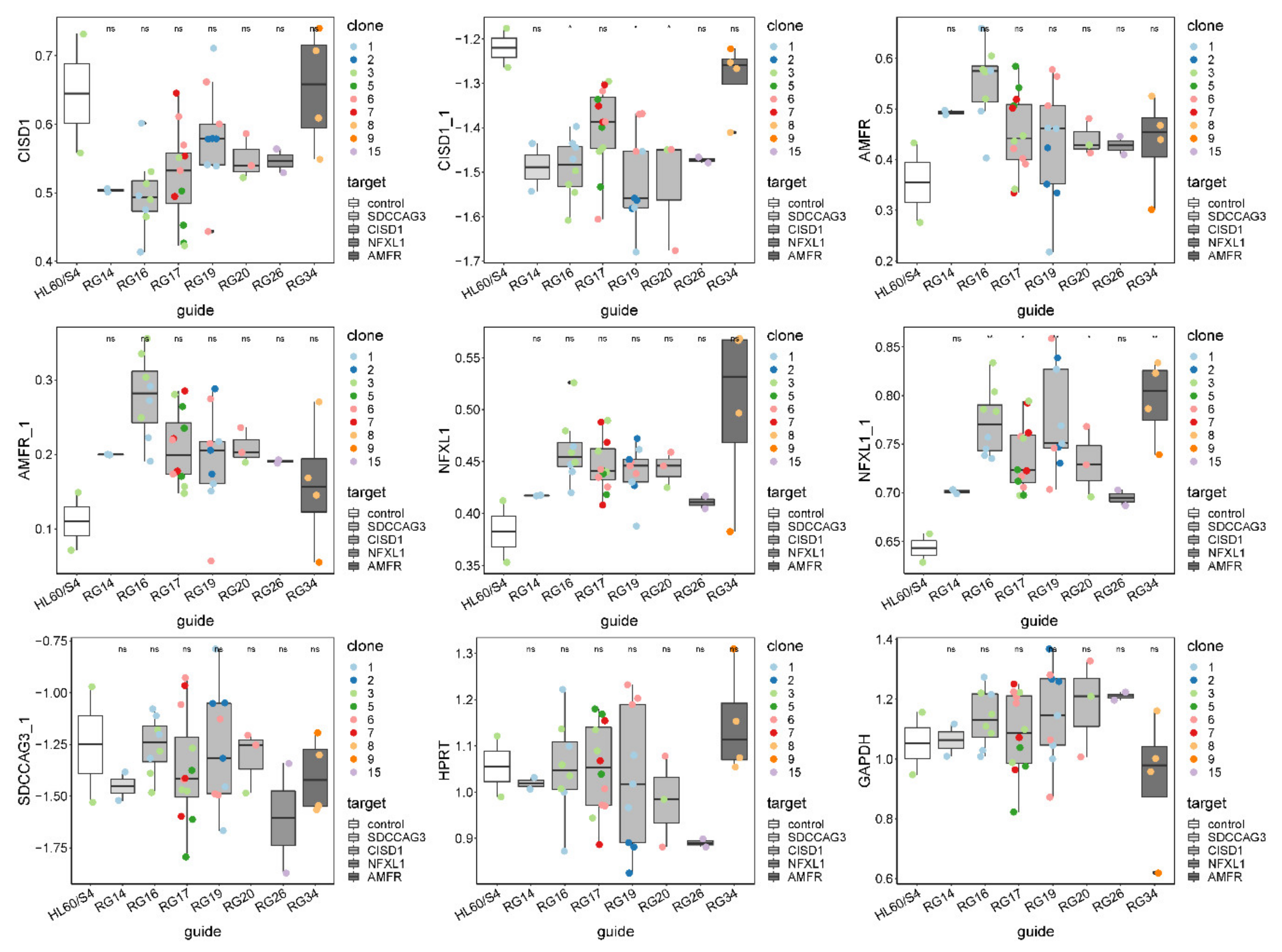
3.2. Isolation and Evaluation of CRISPR-Edited Single-Cell Clones
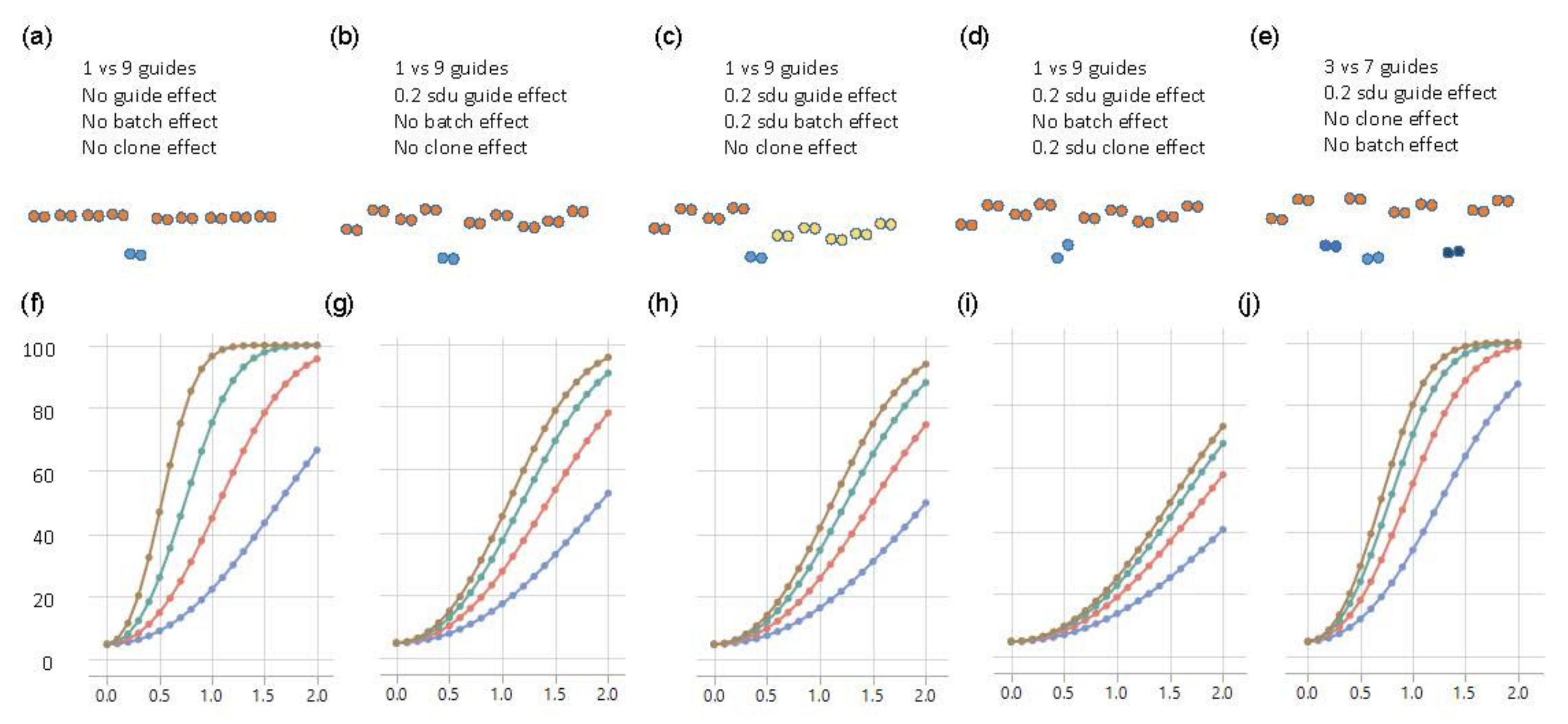
3.3. Simulation Studies to Establish Power of Fluidigm-Based Single-Cell Regulatory Assessment
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Altshuler, D.; Daly, M.J.; Lander, E.S. Genetic mapping in human disease. Science 2008, 322, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Fang, M.; Jostins, L.; Mirkov, M.U.; Boucher, G.; Anderson, C.A.; Andersen, V.; Cleynen, I.; Cortes, A.; Crins, F. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 2017, 547, 173. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, A.; Taliun, D.; Thurner, M.; Robertson, N.R.; Torres, J.M.; Rayner, N.W.; Payne, A.J.; Steinthorsdottir, V.; Scott, R.A.; Grarup, N. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 2018, 50, 1505. [Google Scholar] [CrossRef]
- Zeng, B.; Lloyd-Jones, L.R.; Montgomery, G.W.; Metspalu, A.; Esko, T.; Franke, L.; Vosa, U.; Claringbould, A.; Brigham, K.L.; Quyyumi, A.A. Comprehensive Multiple eQTL Detection and Its Application to GWAS Interpretation. Genetics 2019, 212, 302091. [Google Scholar] [CrossRef]
- Gusev, A.; Lee, S.H.; Trynka, G.; Finucane, H.; Vilhjálmsson, B.J.; Xu, H.; Zang, C.; Ripke, S.; Bulik-Sullivan, B.; Stahl, E. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am. J. Hum. Genet. 2014, 95, 535–552. [Google Scholar] [CrossRef]
- Bulik-Sullivan, B.; Finucane, H.K.; Anttila, V.; Gusev, A.; Day, F.R.; Loh, P.-R.; Duncan, L.; Perry, J.R.; Patterson, N.; Robinson, E.B. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 2015, 47, 1236. [Google Scholar] [CrossRef]
- Li, M.J.; Pan, Z.; Liu, Z.; Wu, J.; Wang, P.; Zhu, Y.; Xu, F.; Xia, Z.; Sham, P.C.; Kocher, J.-P.A. Predicting regulatory variants with composite statistic. Bioinformatics 2016, 32, 2729–2736. [Google Scholar] [CrossRef]
- Liu, L.; Sanderford, M.D.; Patel, R.; Chandrashekar, P.; Gibson, G.; Kumar, S. Biological relevance of computationally predicted pathogenicity of noncoding variants. Nat. Commun. 2019, 10, 330. [Google Scholar] [CrossRef]
- Huo, Y.; Li, S.; Liu, J.; Li, X.; Luo, X.-J. Functional genomics reveal gene regulatory mechanisms underlying schizophrenia risk. Nat. Commun. 2019, 10, 670. [Google Scholar] [CrossRef] [PubMed]
- Gasperini, M.; Hill, A.J.; McFaline-Figueroa, J.L.; Martin, B.; Kim, S.; Zhang, M.D.; Jackson, D.; Leith, A.; Schreiber, J.; Noble, W.S. A Genome-wide framework for mapping gene regulation via cellular genetic screens. Cell 2019, 176, 377–390.e19. [Google Scholar] [CrossRef] [PubMed]
- Adamson, B.; Norman, T.M.; Jost, M.; Cho, M.Y.; Nuñez, J.K.; Chen, Y.; Villalta, J.E.; Gilbert, L.A.; Horlbeck, M.A.; Hein, M.Y. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 2016, 167, 1867–1882.e21. [Google Scholar] [CrossRef] [PubMed]
- Dixit, A.; Parnas, O.; Li, B.; Chen, J.; Fulco, C.P.; Jerby-Arnon, L.; Marjanovic, N.D.; Dionne, D.; Burks, T.; Raychowdhury, R. Perturb-Seq: Dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 2016, 167, 1853–1866.e17. [Google Scholar] [CrossRef]
- Jaitin, D.A.; Weiner, A.; Yofe, I.; Lara-Astiaso, D.; Keren-Shaul, H.; David, E.; Salame, T.M.; Tanay, A.; van Oudenaarden, A.; Amit, I. Dissecting immune circuits by linking CRISPR-pooled screens with single-cell RNA-seq. Cell 2016, 167, 1883–1896.e15. [Google Scholar] [CrossRef]
- Datlinger, P.; Rendeiro, A.F.; Schmidl, C.; Krausgruber, T.; Traxler, P.; Klughammer, J.; Schuster, L.C.; Kuchler, A.; Alpar, D.; Bock, C. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 2017, 14, 297. [Google Scholar] [CrossRef]
- Anderson, C.A.; Boucher, G.; Lees, C.W.; Franke, A.; D’Amato, M.; Taylor, K.D.; Lee, J.C.; Goyette, P.; Imielinski, M.; Latiano, A. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011, 43, 246. [Google Scholar] [CrossRef]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, L.; Wang, C.; Sheng, G.; Feng, L.; Yin, C. Autocrine motility factor receptor promotes the proliferation of human acute monocytic leukemia THP-1 cells. Int. J. Mol. Med. 2015, 36, 627–632. [Google Scholar] [CrossRef]
- Czimmerer, Z.; Varga, T.; Poliska, S.; Nemet, I.; Szanto, A.; Nagy, L. Identification of novel markers of alternative activation and potential endogenous PPARγ ligand production mechanisms in human IL-4 stimulated differentiating macrophages. Immunobiology 2012, 217, 1301–1314. [Google Scholar] [CrossRef]
- Marigorta, U.M.; Denson, L.A.; Hyams, J.S.; Mondal, K.; Prince, J.; Walters, T.D.; Griffiths, A.; Noe, J.D.; Crandall, W.V.; Rosh, J.R.; et al. Transcriptional risk scores link GWAS to eQTLs and predict complications in Crohn’s disease. Nat. Genet. 2017, 49, 1517–1521. [Google Scholar] [CrossRef]
- Lloyd-Jones, L.R.; Holloway, A.; McRae, A.; Yang, J.; Small, K.; Zhao, J.; Zeng, B.; Bakshi, A.; Metspalu, A.; Dermitzakis, M. The genetic architecture of gene expression in peripheral blood. Am. J. Hum. Genet. 2017, 100, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Fairfax, B.P.; Humburg, P.; Makino, S.; Narambhai, V.; Wong, D.; Laou, E.; Jostins, L.; Plant, K.; Andrews, R.; McGee, C.; et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 2014, 343, 1246949. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Tamura, K.; Sanderford, M.; Gray, V.E.; Kumar, S. A molecular evolutionary reference for the human variome. Mol. Biol. Evol. 2015, 33, 245–254. [Google Scholar] [CrossRef]
- Westra, H.J.; Peters, M.J.; Esko, T.; Yaghootkar, H.; Schurmann, C.; Kettunen, J.; Christiansen, M.W.; Fairfax, B.P.; Schramm, K.; Powell, J.E.; et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 2013, 45, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2006, 35, D61–D65. [Google Scholar] [CrossRef] [PubMed]
- Cradick, T.J.; Qiu, P.; Lee, C.M.; Fine, E.J.; Bao, G. COSMID: A web-based tool for identifying and validating CRISPR/Cas off-target sites. Mol. Ther. Nucleic Acids 2014, 3, e214. [Google Scholar] [CrossRef]
- Breitman, T.; Selonick, S.E.; Collins, S.J. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc. Natl. Acad. Sci. USA 1980, 77, 2936–2940. [Google Scholar] [CrossRef]
- Okazaki, T.; Bell, R.M.; Hannun, Y.A. Sphingomyelin turnover induced by vitamin D3 in HL-60 cells. Role in cell differentiation. J. Biol. Chem. 1989, 264, 19076–19080. [Google Scholar] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J. From FastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.11–11.10.33. [Google Scholar]
- Preininger, M.; Arafat, D.; Kim, J.; Nath, A.P.; Idaghdour, Y.; Brigham, K.L.; Gibson, G. Blood-informative transcripts define nine common axes of peripheral blood gene expression. PLoS Genet. 2013, 9, e1003362. [Google Scholar] [CrossRef]
- Jang, J.S.; Simon, V.A.; Feddersen, R.M.; Rakhshan, F.; Schultz, D.A.; Zschunke, M.A.; Lingle, W.L.; Kolbert, C.P.; Jen, J. Quantitative miRNA expression analysis using fluidigm microfluidics dynamic arrays. BMC Genom. 2011, 12, 144. [Google Scholar] [CrossRef] [PubMed]
- Coudray-Meunier, C.; Fraisse, A.; Martin-Latil, S.; Delannoy, S.; Fach, P.; Perelle, S. A novel high-throughput method for molecular detection of human pathogenic viruses using a nanofluidic real-time PCR System. PLoS ONE 2016, 11, e0147832. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Freire, V.; Ebert, A.D.; Kalisky, T.; Quake, S.R.; Wu, J.C. Microfluidic single-cell real-time PCR for comparative analysis of gene expression patterns. Nat. Protoc. 2012, 7, 829–838. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.J.; Gallo, R.C.; Gallagher, R.E. Continuous growth and differentiation of human myeloid leukaemic cells in suspension culture. Nature 1977, 270, 347. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, R.; Collins, S.; Trujillo, J.; McCredie, K.; Ahearn, M.; Tsai, S.; Metzgar, R.; Aulakh, G.; Ting, R.; Ruscetti, F. Characterization of the continuous, differentiating myeloid cell line (HL-60) from a patient with acute promyelocytic leukemia. Blood 1979, 54, 713–733. [Google Scholar] [CrossRef]
- Leung, M.-F.; Sokoloski, J.A.; Sartorelli, A.C. Changes in microtubules, microtubule-associated proteins, and intermediate filaments during the differentiation of HL-60 leukemia cells. Cancer Res. 1992, 52, 949–954. [Google Scholar]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325. [Google Scholar] [CrossRef]
- Ramirez, R.N.; El-Ali, N.C.; Mager, M.A.; Wyman, D.; Conesa, A.; Mortazavi, A. Dynamic gene regulatory networks of human myeloid differentiation. Cell Syst. 2017, 4, 416–429.e3. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2018, 47, D886–D894. [Google Scholar] [CrossRef]
- Korkmaz, G.; Lopes, R.; Ugalde, A.P.; Nevedomskaya, E.; Han, R.; Myacheva, K.; Zwart, W.; Elkon, R.; Agami, R. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 192. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, N.; Srinivasan, S.; Kooshesh, K.; Guo, Y.; Edwards, M.D.; Banerjee, B.; Syed, T.; Emons, B.J.; Gifford, D.K.; Sherwood, R.I. High-throughput mapping of regulatory DNA. Nat. Biotechnol. 2016, 34, 167. [Google Scholar] [CrossRef] [PubMed]
- Simeonov, D.R.; Gowen, B.G.; Boontanrart, M.; Roth, T.L.; Gagnon, J.D.; Mumbach, M.R.; Satpathy, A.T.; Lee, Y.; Bray, N.L.; Chan, A.Y. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature 2017, 549, 111. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Tian, R.; Lee, C.M.; Bao, G.; Gibson, G. Fine-mapping within eQTL Credible Intervals by Expression CROP-seq. Biol. Meth. Protoc. 2020. [Google Scholar] [CrossRef]
- Tewhey, R.; Kotliar, D.; Park, D.S.; Liu, B.; Winnicki, S.; Reilly, S.K.; Andersen, K.G.; Mikkelsen, T.S.; Lander, E.S.; Schaffner, S.F. Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell 2016, 165, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Kreimer, A.; Zeng, H.; Edwards, M.D.; Guo, Y.; Tian, K.; Shin, S.; Welch, R.; Wainberg, M.; Mohan, R.; Sinnott-Armstrong, N.A. Predicting gene expression in massively parallel reporter assays: A comparative study. Hum. Mutat. 2017, 38, 1240–1250. [Google Scholar] [CrossRef]
- Kalita, C.A.; Brown, C.D.; Freiman, A.; Isherwood, J.; Wen, X.; Pique-Regi, R.; Luca, F. High-throughput characterization of genetic effects on DNA–protein binding and gene transcription. Genome Res. 2018, 28, 1701–1708. [Google Scholar] [CrossRef]
- Norga, K.K.; Gurganus, M.C.; Dilda, C.L.; Yamamoto, A.; Lyman, R.F.; Patel, P.H.; Rubin, G.M.; Hoskins, R.A.; Mackay, T.F.; Bellen, H.J. Quantitative analysis of bristle number in Drosophila mutants identifies genes involved in neural development. Curr. Biol. 2003, 13, 1388–1396. [Google Scholar] [CrossRef]
- Magwire, M.M.; Yamamoto, A.; Carbone, M.A.; Roshina, N.V.; Symonenko, A.V.; Pasyukova, E.G.; Morozova, T.V.; Mackay, T.F. Quantitative and molecular genetic analyses of mutations increasing Drosophila life span. PLoS Genet. 2010, 6, e1001037. [Google Scholar] [CrossRef]
- Metzger, B.P.; Yuan, D.C.; Gruber, J.D.; Duveau, F.; Wittkopp, P.J. Selection on noise constrains variation in a eukaryotic promoter. Nature 2015, 521, 344. [Google Scholar] [CrossRef]
- Li, X.; Kim, Y.; Tsang, E.K.; Davis, J.R.; Damani, F.N.; Chiang, C.; Hess, G.T.; Zappala, Z.; Strober, B.J.; Scott, A.J. The impact of rare variation on gene expression across tissues. Nature 2017, 550, 239. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Akinsanmi, I.; Arafat, D.; Cradick, T.; Lee, C.M.; Banskota, S.; Marigorta, U.M.; Bao, G.; Gibson, G. A burden of rare variants associated with extremes of gene expression in human peripheral blood. Am. J. Hum. Genet. 2016, 98, 299–309. [Google Scholar] [CrossRef] [PubMed]
- van Overbeek, M.; Capurso, D.; Carter, M.M.; Thompson, M.S.; Frias, E.; Russ, C.; Reece-Hoyes, J.S.; Nye, C.; Gradia, S.; Vidal, B. DNA repair profiling reveals nonrandom outcomes at Cas9-mediated breaks. Mol. Cell 2016, 63, 633–646. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| gRNA | Gene | Top SNP | SNP | Z-Score | Type | Genome Location | Coding Region |
|---|---|---|---|---|---|---|---|
| RG14 | SDCCAG3 | rs10870171 | rs3812594 | −34.60 | High CADD | Exon of SEC16A | Yes |
| RG16 | CISD1 | rs4397793 | rs4397793 | −23.84 | Top | Intron of TFAM | No |
| RG17 | CISD1 | rs4397793 | rs648138 | −70.54 | Control | Intergenic of TFAM | No |
| RG19 | CISD1 | rs2590375 | rs2590363 | −100.37 | Both | Intron of IPMK | No |
| RG20 | CISD1 | rs2590375 | rs1416763 | −100.27 | Both | Intron of CISD1 | No |
| RG26 | NFXL1 | rs116521751 | rs321622 | −63.35 | Both | Intron of NIPAL1 | No |
| RG34 | AMFR | rs8060037 | rs8060037 | −14.09 | Top | Intron of NUDT21 | No |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, R.; Pan, Y.; Etheridge, T.H.A.; Deshmukh, H.; Gulick, D.; Gibson, G.; Bao, G.; Lee, C.M. Pitfalls in Single Clone CRISPR-Cas9 Mutagenesis to Fine-Map Regulatory Intervals. Genes 2020, 11, 504. https://doi.org/10.3390/genes11050504
Tian R, Pan Y, Etheridge THA, Deshmukh H, Gulick D, Gibson G, Bao G, Lee CM. Pitfalls in Single Clone CRISPR-Cas9 Mutagenesis to Fine-Map Regulatory Intervals. Genes. 2020; 11(5):504. https://doi.org/10.3390/genes11050504
Chicago/Turabian StyleTian, Ruoyu, Yidan Pan, Thomas H. A. Etheridge, Harshavardhan Deshmukh, Dalia Gulick, Greg Gibson, Gang Bao, and Ciaran M Lee. 2020. "Pitfalls in Single Clone CRISPR-Cas9 Mutagenesis to Fine-Map Regulatory Intervals" Genes 11, no. 5: 504. https://doi.org/10.3390/genes11050504
APA StyleTian, R., Pan, Y., Etheridge, T. H. A., Deshmukh, H., Gulick, D., Gibson, G., Bao, G., & Lee, C. M. (2020). Pitfalls in Single Clone CRISPR-Cas9 Mutagenesis to Fine-Map Regulatory Intervals. Genes, 11(5), 504. https://doi.org/10.3390/genes11050504