Pathogenic Effect of GDAP1 Gene Mutations in a Yeast Model


Abstract
1. Introduction
2. Materials and Methods
2.1. Strains, Media, and Growth Conditions
2.2. Plasmids
2.3. Site Directed Mutagenesis
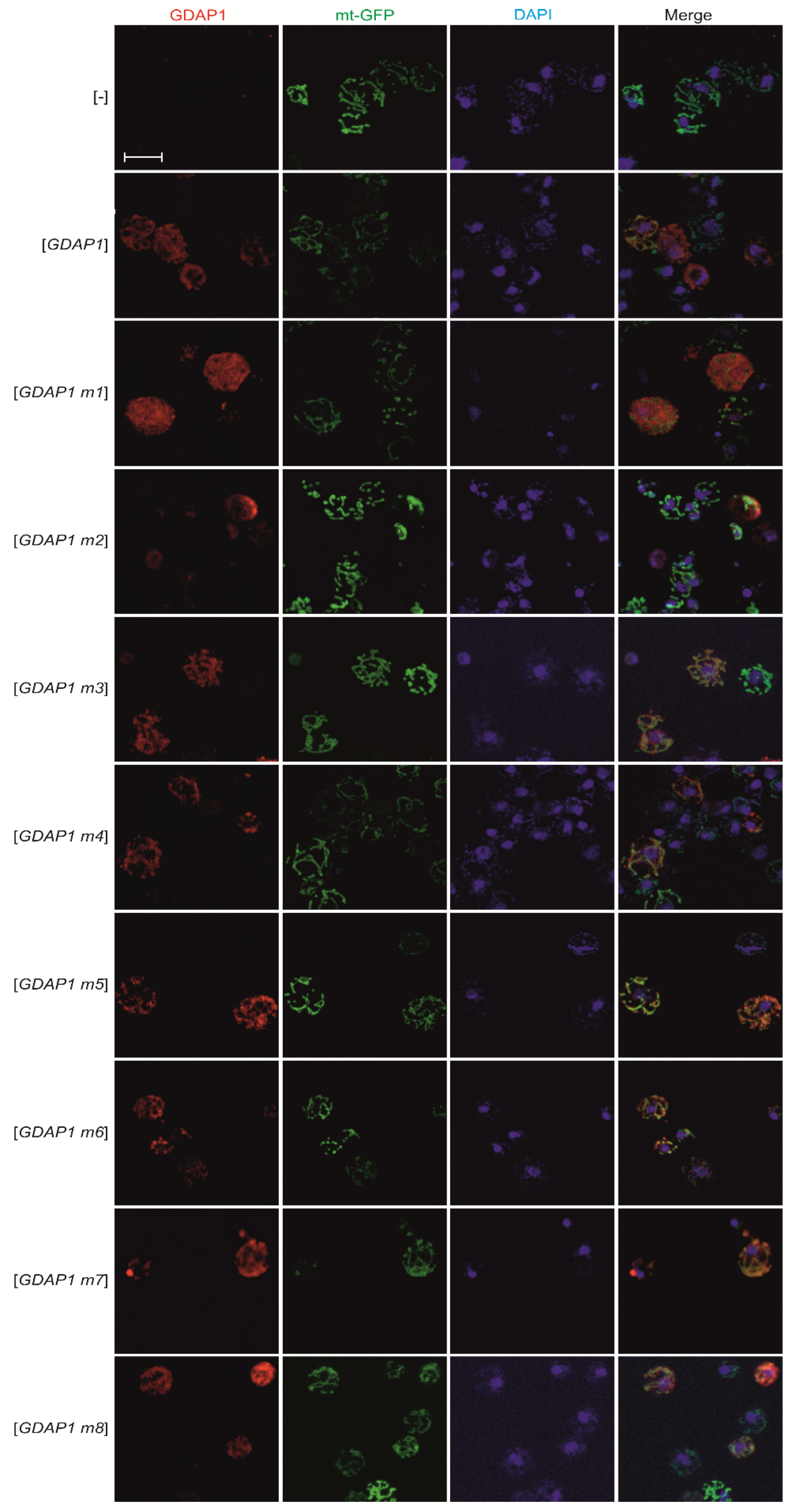
2.4. Fluorescence Microscopy
2.5. Confocal Microscopy
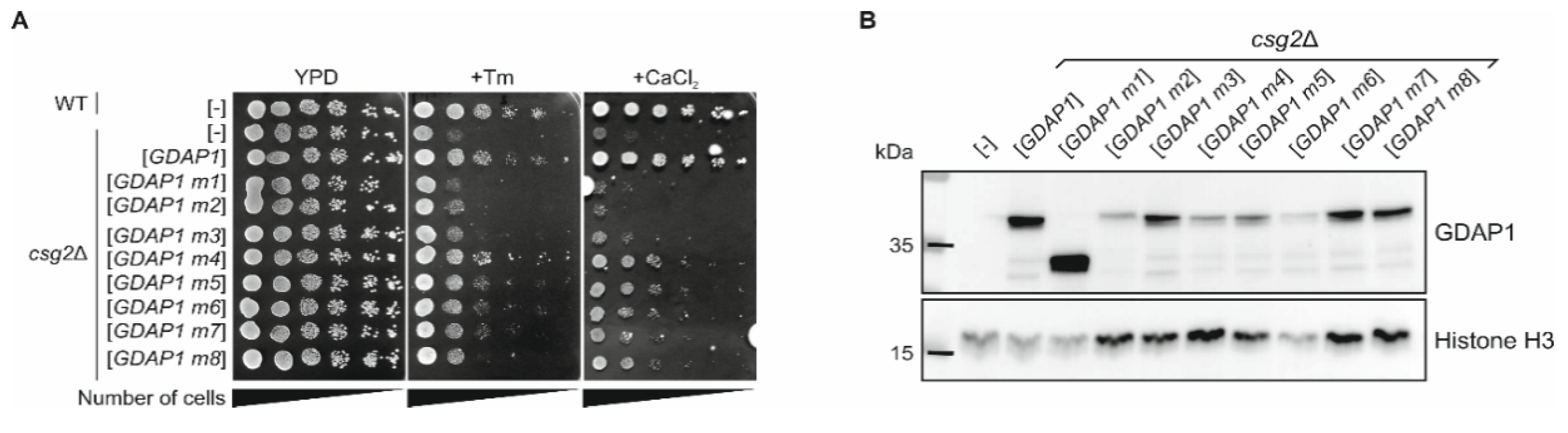
2.6. Western Blot Analysis
2.7. DNA Escape Assay
2.8. Estimation of Yeast Respiratory-Deficiency
3. Results
3.1. Selection of GDAP1 Mutations
3.2. cDNA of the Human GDAP1 Gene Coding for Ganglioside Induced Differentiation Associated Protein 1 Is Stable When Heterologously Expressed in Yeast Cells
3.3. Specific GDAP1 Mutations Cause Mislocalization of the Resulting GDAP1 Protein
3.4. Expression of Human GDAP1 cDNA Affects Mitochondrial Network Formation in Yeast
3.5. Some GDAP1 Protein Mutants When Produced in Yeast Increase the Rate of Mitochondrial DNA Escape to the Nucleus
3.6. The Expression of Human Wild-Type GDAP1 Gene Reduces the Growth Defect of the csg2 Mutant Yeast Strain
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence | |
|---|---|---|
| GDAP1 m2: c.980G>A; p.Gly327Asp | GDAP1m2F | 5′-cactcctgcaagcaaatcaaccacaagggtcgt-3′ |
| GDAP1m2R | 5′-acgacccttgtggttgatttgcttgcaggagtg-3′ | |
| GDAP1 m3: c.652C>G p.Gln218Glu | GDAP1m3F | 5′-caattcagtttcaacctcatccaagactttctccaactc-3′ |
| GDAP1m3R | 5′-gagttggagaaagtcttggatgaggttgaaactgaattg-3′ | |
| GDAP1 m4: c.664G>A p.Glu222Lys | GDAP1m4F | 5′-ttcttcatttcttctttgcaatttagtttcaacctgatccaagactt-3′ |
| GDAP1m4R | 5′-agtcttggatcaggttgaaactaaattgcaaagaagaaatgaagaa-3′ | |
| GDAP1 m5: c.715C>T p.Leu239Phe | GDAP1m5F | 5′-ggattcaccgcagaaccaaggttgctggc-3′ |
| GDAP1m5R | 5′-gccagcaaccttggttctgcggtgaatcc-3′ | |
| GDAP1 m6: c.368A>G p.His123Arg | GDAP1m6F | 5′-gcagctctcggtaacgttgtacccgtggg-3′ |
| GDAP1m6R | 5′-cccacgggtacaacgttaccgagagctgc-3′ | |
| GDAP1 m7: c.347T>C p.Met116Thr | GDAP1m7F | 5′-gtacccgtgggtaatacgtgctttctttatcaggca-3′ |
| GDAP1m7R | 5′-tgcctgataaagaaagcacgtattacccacgggtac-3′ | |
| GDAP1 m8: c.467C>G p.Ala156Gly | GDAP1m8F GDAP1m8R | 5′-tacgaatccttgtagttccataagccgggatcatg-3′ 5′-catgatcccggcttatggaactacaaggattcgta-3′ |

References
- Lupski, J.R.; Belmont, J.W.; Boerwinkle, E.; Gibbs, R.A. Clan Genomics and the Complex Architecture of Human Disease. Cell 2011, 147, 32–43. [Google Scholar] [CrossRef]
- Gonzaga-Jauregui, C.; Harel, T.; Gambin, T.; Kousi, M.; Griffin, L.B.; Francescatto, L.; Ozes, B.; Karaca, E.; Jhangiani, S.N.; Bainbridge, M.N.; et al. Exome Sequence Analysis Suggests that Genetic Burden Contributes to Phenotypic Variability and Complex Neuropathy. Cell Rep. 2015, 12, 1169–1183. [Google Scholar] [CrossRef]
- Baxter, R.V.; Ben Othmane, K.; Rochelle, J.M.; Stajich, J.E.; Hulette, C.; Dew-Knight, S.; Hentati, F.; Ben Hamida, M.; Bel, S.; Stenger, J.E.; et al. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat. Genet. 2002, 30, 21–22. [Google Scholar] [CrossRef]
- Cuesta, A.; Pedrola, L.; Sevilla, T.; García-Planells, J.; Chumillas, M.J.; Mayordomo, F.; LeGuern, E.; Marín, I.; Vílchez, J.J.; Palau, F. The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat. Genet. 2002, 30, 22–25. [Google Scholar] [CrossRef]
- González-Sánchez, P.; Satrústegui, J.; Palau, F.; del Arco, A. Calcium Deregulation and Mitochondrial Bioenergetics in GDAP1-Related CMT Disease. Int. J. Mol. Sci. 2019, 20, 403. [Google Scholar] [CrossRef] [PubMed]
- Rzepnikowska, W.; Kochański, A. A role for the GDAP1 gene in the molecular pathogenesis of Charcot-Marie-Tooth disease. Acta Neurobiol. Exp. (Wars) 2018, 78, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Azzedine, H.; Ruberg, M.; Ente, D.; Gilardeau, C.; Périé, S.; Wechsler, B.; Brice, A.; LeGuern, E.; Dubourg, O. Variability of disease progression in a family with autosomal recessive CMT associated with a S194X and new R310Q mutation in the GDAP1 gene. Neuromuscul. Disord. 2003, 13, 341–346. [Google Scholar] [CrossRef]
- Claramunt, R.; Pedrola, L.; Sevilla, T.; López de Munain, A.; Berciano, J.; Cuesta, A.; Sánchez-Navarro, B.; Millán, J.M.; Saifi, G.M.; Lupski, J.R.; et al. Genetics of Charcot-Marie-Tooth disease type 4A: Mutations, inheritance, phenotypic variability, and founder effect. J. Med. Genet. 2005, 42, 358–365. [Google Scholar] [CrossRef]
- Liu, H.; Nakagawa, T.; Kanematsu, T.; Uchida, T.; Tsuji, S. Isolation of 10 differentially expressed cDNAs in differentiated Neuro2a cells induced through controlled expression of the GD3 synthase gene. J. Neurochem. 1999, 72, 1781–1790. [Google Scholar] [CrossRef]
- Wagner, K.M.; Rüegg, M.; Niemann, A.; Suter, U. Targeting and Function of the Mitochondrial Fission Factor GDAP1 Are Dependent on Its Tail-Anchor. PLoS ONE 2009, 4, e5160. [Google Scholar] [CrossRef]
- Pedrola, L.; Espert, A.; Wu, X.; Claramunt, R.; Shy, M.E.; Palau, F. GDAP1, the protein causing Charcot-Marie-Tooth disease type 4A, is expressed in neurons and is associated with mitochondria. Hum. Mol. Genet. 2005, 14, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Pedrola, L.; Espert, A.; Valdés-Sánchez, T.; Sánchez-Piris, M.; Sirkowski, E.E.; Scherer, S.S.; Fariñas, I.; Palau, F. Cell expression of GDAP1 in the nervous system and pathogenesis of Charcot-Marie-Tooth type 4A disease. J. Cell. Mol. Med. 2008, 12, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Niemann, A.; Ruegg, M.; La Padula, V.; Schenone, A.; Suter, U. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network. J. Cell Biol. 2005, 170, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Niemann, A.; Wagner, K.M.; Ruegg, M.; Suter, U. GDAP1 mutations differ in their effects on mitochondrial dynamics and apoptosis depending on the mode of inheritance. Neurobiol. Dis. 2009, 36, 509–520. [Google Scholar] [CrossRef] [PubMed]
- González-Sánchez, P.; Pla-Martín, D.; Martínez-Valero, P.; Rueda, C.B.; Calpena, E.; Del Arco, A.; Palau, F.; Satrústegui, J. CMT-linked loss-of-function mutations in GDAP1 impair store-operated Ca(2+) entry-stimulated respiration. Sci. Rep. 2017, 7, 42993. [Google Scholar] [CrossRef] [PubMed]
- Pla-Martín, D.; Rueda, C.B.; Estela, A.; Sánchez-Piris, M.; González-Sánchez, P.; Traba, J.; de la Fuente, S.; Scorrano, L.; Renau-Piqueras, J.; Alvarez, J.; et al. Silencing of the Charcot-Marie-Tooth disease-associated gene GDAP1 induces abnormal mitochondrial distribution and affects Ca2+ homeostasis by reducing store-operated Ca2+ entry. Neurobiol. Dis. 2013, 55, 140–151. [Google Scholar] [CrossRef]
- Niemann, A.; Huber, N.; Wagner, K.M.; Somandin, C.; Horn, M.; Lebrun-Julien, F.; Angst, B.; Pereira, J.A.; Halfter, H.; Welzl, H.; et al. The Gdap1 knockout mouse mechanistically links redox control to Charcot-Marie-Tooth disease. Brain 2014, 137, 668–682. [Google Scholar] [CrossRef]
- Noack, R.; Frede, S.; Albrecht, P.; Henke, N.; Pfeiffer, A.; Knoll, K.; Dehmel, T.; Meyer zu Horste, G.; Stettner, M.; Kieseier, B.C.; et al. Charcot-Marie-Tooth disease CMT4A: GDAP1 increases cellular glutathione and the mitochondrial membrane potential. Hum. Mol. Genet. 2012, 21, 150–162. [Google Scholar] [CrossRef]
- Estela, A.; Pla-Martín, D.; Sánchez-Piris, M.; Sesaki, H.; Palau, F. Charcot-Marie-Tooth-related gene GDAP1 complements cell cycle delay at G2/M phase in Saccharomyces cerevisiae fis1 gene-defective cells. J. Biol. Chem. 2011, 286, 36777–36786. [Google Scholar] [CrossRef]
- Thorsness, P.E.; Fox, T.D. Nuclear mutations in Saccharomyces cerevisiae that affect the escape of DNA from mitochondria to the nucleus. Genetics 1993, 134, 21–28. [Google Scholar]
- Mumberg, D.; Müller, R.; Funk, M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 1995, 156, 119–122. [Google Scholar] [CrossRef]
- Westermann, B.; Neupert, W. Mitochondria-targeted green fluorescent proteins: Convenient tools for the study of organelle biogenesis in Saccharomyces cerevisiae. Yeast 2000, 16, 1421–1427. [Google Scholar] [CrossRef]
- Zimon, M.; Baets, J.; Fabrizi, G.M.; Jaakkola, E.; Kabzinska, D.; Pilch, J.; Schindler, A.B.; Cornblath, D.R.; Fischbeck, K.H.; Auer-Grumbach, M.; et al. Dominant GDAP1 mutations cause predominantly mild CMT phenotypes. Neurology 2011, 77, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Kabzińska, D.; Strugalska-Cynowska, H.; Kostera-Pruszczyk, A.; Ryniewicz, B.; Posmyk, R.; Midro, A.; Seeman, P.; Báranková, L.; Zimoń, M.; Baets, J.; et al. L239F founder mutation in GDAP1 is associated with a mild Charcot–Marie–Tooth type 4C4 (CMT4C4) phenotype. Neurogenetics 2010, 11, 357–366. [Google Scholar] [CrossRef]
- Kabzińska, D.; Niemann, A.; Drac, H.; Huber, N.; Potulska-Chromik, A.; Hausmanowa-Petrusewicz, I.; Suter, U.; Kochański, A. A new missense GDAP1 mutation disturbing targeting to the mitochondrial membrane causes a severe form of AR-CMT2C disease. Neurogenetics 2011, 12, 145–153. [Google Scholar] [CrossRef]
- Kabzińska, D.; Kotruchow, K.; Cegielska, J.; Hausmanowa-Petrusewicz, I.; Kochański, A. A severe recessive and a mild dominant form of Charcot-Marie-Tooth disease associated with a newly identified Glu222Lys GDAP1 gene mutation. Acta Biochim. Pol. 2014, 61, 739–744. [Google Scholar] [CrossRef]
- Lewis, S.C.; Uchiyama, L.F.; Nunnari, J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 2016, 353, aaf5549. [Google Scholar] [CrossRef]
- Murley, A.; Lackner, L.L.; Osman, C.; West, M.; Voeltz, G.K.; Walter, P.; Nunnari, J. ER-associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast. eLife 2013, 2, e00422. [Google Scholar] [CrossRef]
- Campbell, C.L.; Thorsness, P.E. Escape of mitochondrial DNA to the nucleus in yme1 yeast is mediated by vacuolar-dependent turnover of abnormal mitochondrial compartments. J. Cell Sci. 1998, 111 Pt 1, 2455–2464. [Google Scholar]
- Thorsness, M.K.; White, K.H.; Thorsness, P.E. Migration of mtDNA into the Nucleus. In Mitochondrial DNA.; Humana Press: New Jersey, NJ, USA, 2002; pp. 177–186. [Google Scholar]
- Shafer, K.S.; Hanekamp, T.; White, K.H.; Thorsness, P.E. Mechanisms of mitochondrial DNA escape to the nucleus in the yeast Saccharomyces cerevisiae. Curr. Genet. 1999, 36, 183–194. [Google Scholar] [CrossRef]
- Xu, N.; Thorsness, M.K.; Thorsness, P.E. Mitochondrial DNA impacts the morphology of mitochondrial compartments. Gene 2005, 354, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Knupp, J.; Martinez-Montañés, F.; Van Den Bergh, F.; Cottier, S.; Schneiter, R.; Beard, D.; Chang, A. Sphingolipid accumulation causes mitochondrial dysregulation and cell death. Cell Death Differ. 2017, 24, 2044–2053. [Google Scholar] [CrossRef] [PubMed]




| Plasmid | Source or Reference |
|---|---|
| pCMV6-XL5-GDAP1 | OriGene |
| p425-PTDH3 [2µ; LEU2] | [21] |
| p425-PTDH3-GDAP1 | This study |
| p425-PTDH3-GDAP1m1 | This study |
| p425-PTDH3-GDAP1m2 | This study |
| p425-PTDH3-GDAP1m3 | This study |
| p425-PTDH3-GDAP1m4 | This study |
| p425-PTDH3-GDAP1m5 | This study |
| p425-PTDH3-GDAP1m6 | This study |
| p425-PTDH3-GDAP1m7 | This study |
| p425-PTDH3-GDAP1m8 | This study |
| PXY122 (mt-GFP) [2µ, HIS3] | [22] |
| GDAP1 Variant (aa Substitution) | Localization | Inheritance of GDAP1 Mutation | Petite | mtDNA Escape | Effect on csg2Δ | Summarized Effect in Yeast | Clinical Phenotype | |
|---|---|---|---|---|---|---|---|---|
| Tm | CaCl2 | |||||||
| GDAP1 | M | _ | 12 | - | + | + | _ | _ |
| GDAP1 m1 (ΔC-terminus) | C | AR | 4 | - | - | - | strong | severe |
| GDAP1 m2 (Gly327Asp) | C | AR | 5 | moderate | - | - | strong | severe |
| GDAP1 m3 (Glu218Glu) | M | AR | 11 | - | - | - | strong/ moderate | moderate to severe |
| GDAP1 m4 (Glu222Lys) | M | AR/AD | 9 | minimal | + | + | weak | weak to moderate |
| GDAP1 m5 (Leu239Phe) | M | AR | 10 | maximal | -/+ | -/+ | moderate | moderate in homozygote, lack in heterozygote |
| GDAP1 m6 (His123Arg) | M | AD | 5 | - | -/+ | -/+ | weak | weak |
| GDAP1 m7 (Met116Thr) | M | AR | 5 | - | -/+ | -/+ | weak | moderate in homozygote |
| GDAP1 m8 (Ala156Gly) | M | AD | 6 | - | - | -/+ | moderate | moderate in heterozygote |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rzepnikowska, W.; Kaminska, J.; Kabzińska, D.; Kochański, A. Pathogenic Effect of GDAP1 Gene Mutations in a Yeast Model. Genes 2020, 11, 310. https://doi.org/10.3390/genes11030310
Rzepnikowska W, Kaminska J, Kabzińska D, Kochański A. Pathogenic Effect of GDAP1 Gene Mutations in a Yeast Model. Genes. 2020; 11(3):310. https://doi.org/10.3390/genes11030310
Chicago/Turabian StyleRzepnikowska, Weronika, Joanna Kaminska, Dagmara Kabzińska, and Andrzej Kochański. 2020. "Pathogenic Effect of GDAP1 Gene Mutations in a Yeast Model" Genes 11, no. 3: 310. https://doi.org/10.3390/genes11030310
APA StyleRzepnikowska, W., Kaminska, J., Kabzińska, D., & Kochański, A. (2020). Pathogenic Effect of GDAP1 Gene Mutations in a Yeast Model. Genes, 11(3), 310. https://doi.org/10.3390/genes11030310