The Effect of Tobacco Smoking Differs across Indices of DNA Methylation-Based Aging in an African American Sample: DNA Methylation-Based Indices of Smoking Capture These Effects

 ,
, 
Abstract
1. Introduction
1.1. Chronological Aging
1.2. Phenotypic (Morbidity) Aging
1.3. GRIM and Its Subscales
1.4. Telomere Length
1.5. Controlling Cell-type Variation
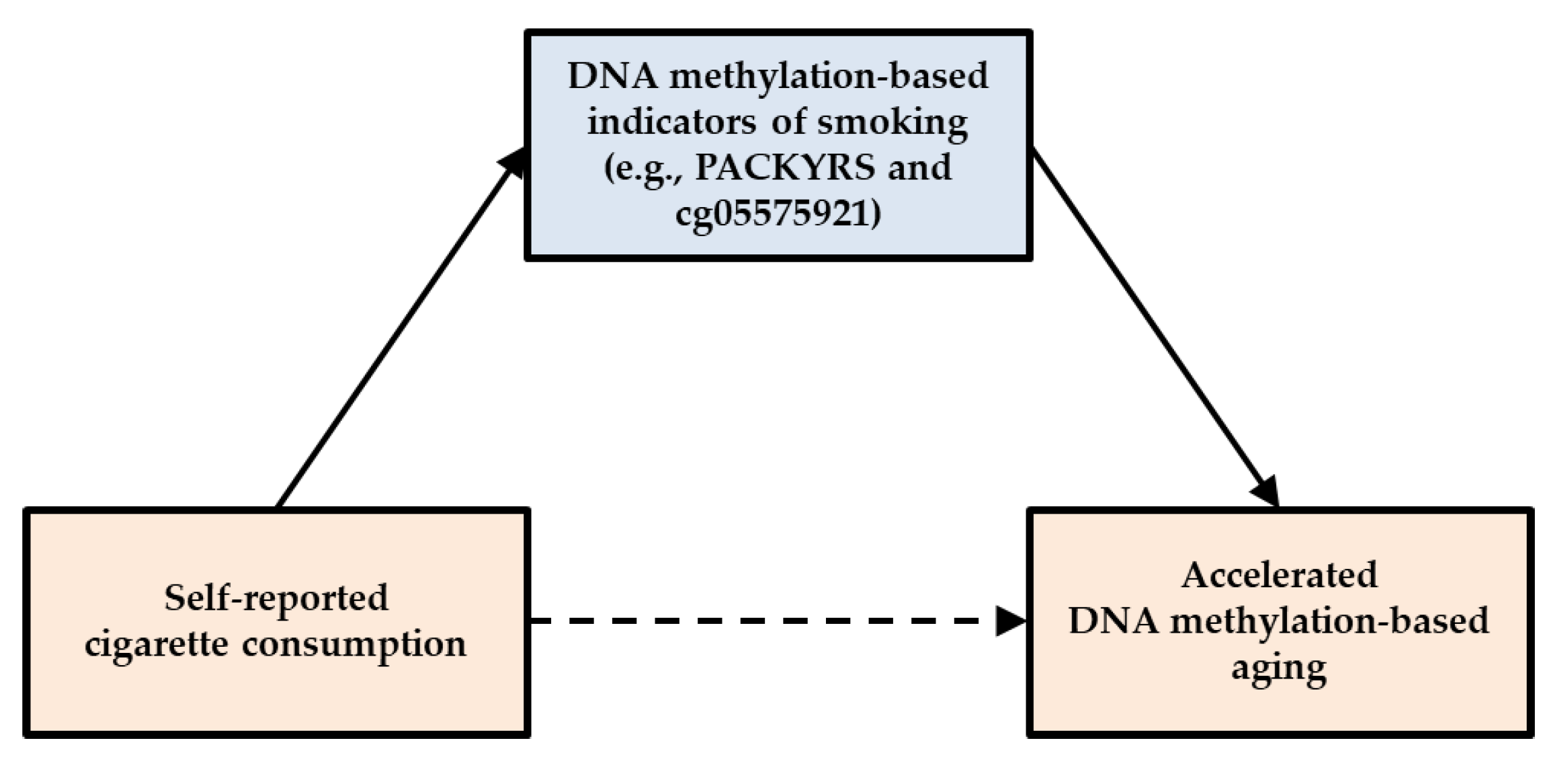
1.6. Specific Hypotheses
2. Materials and Methods
2.1. Sample
2.2. Procedures
2.3. DNA Methylation Procedures
2.4. Measures
2.4.1. Self-reported Cigarette Consumption
2.4.2. Accelerated DNA Methylation-Based Aging
2.4.3. DNA Methylation-Based Estimate of Telomere Length
2.4.4. Components of GrimAge
2.4.5. Transformation to Accelerated Aging
2.4.6. Cell-Type Composition
2.4.7. Control Variables
2.5. Analytic Approach
3. Results
3.1. Initial Findings
3.2. Model Testing
3.3. Testing of Direct and Indirect Effects
3.4. Sensitivity Analysis
4. Conclusions and Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lariscy, J.T. Smoking-attributable mortality by cause of death in the United States: An indirect approach. SSM-Popul. Health 2019, 7, 100349. [Google Scholar] [PubMed]
- Heron, M.; Hoyert, D.; Murphy, S.; Jiaquan, X.; Kochanek, K.; Tejada-Vera, B. Deaths: Final data for 2006. Natl. Vital Stat. Rep. 2009, 57, 1–4. [Google Scholar] [PubMed]
- United States Department of Health and Human Services. Tobacco use among U.S. racial/ethnic minority groups—African Americans, American Indians and Alaska Natives, Asian Americans and Pacific Islanders, and Hispanics: A report of the Surgeon General; United States Department of Health and Human Services, Centers for Disease Control and Prevention: Atlanta, GA, USA, 1998. [Google Scholar]
- Kochanek, K.; Murphy, S.; Xu, J.; Tejada-Vera, B. Deaths: Final data for 2014. Natl. Vital Stat. Rep. 2016, 65, 1–4. [Google Scholar]
- Murphy, S.L.; Xu, J.; Kochanek, K.D. Deaths: Final data for 2010. Natl. Vital Stat. Rep. 2013, 62, 1–4. [Google Scholar]
- Murphy, S.E.; Park, S.L.; Balbo, S.; Haiman, C.A.; Hatsukami, D.K.; Patel, Y.; Peterson, L.A.; Stepanov, I.; Stram, D.O.; Tretyakova, N.; et al. Tobacco biomarkers and genetic/epigenetic analysis to investigate ethnic/racial differences in lung cancer risk among smokers. NPJ Precis. Oncol. 2018, 2, 1–10. [Google Scholar] [CrossRef]
- American Lung Association. Too many cases, too many deaths: Lung cancer in African Americans; American Lung Association: Washington, DC, USA, 2010. [Google Scholar]
- Beach, S.R.H.; Dogan, M.V.; Lei, M.-K.; Cutrona, C.E.; Gerrard, M.; Gibbons, F.X.; Simons, R.L.; Brody, G.H.; Philibert, R.A. Methylomic aging as a window onto the influence of lifestyle: Tobacco and alcohol use alter the rate of biological aging. J. Am. Geriatr. Soc. 2015, 63, 2519–2525. [Google Scholar] [CrossRef]
- Beach, S.R.H.; Gerrard, M.; Gibbons, F.X.; Brody, G.H.; Philibert, R.A. A role for epigenetics in broadening the scope of pediatric care in the prevention of adolescent smoking. Epigenet. Diagn. Ther. 2016, 1, 91–97. [Google Scholar] [CrossRef][Green Version]
- Lei, M.-K.; Beach, S.R.H.; Dogan, M.V.; Philibert, R.A. A Pilot investigation of the impact of smoking cessation on biological age. Am. J. Addict. 2017, 26, 129–135. [Google Scholar] [CrossRef]
- Monick, M.M.; Beach, S.R.H.; Plume, J.; Sears, R.; Gerrard, M.; Brody, G.H.; Philbert, R.A. Coordinated changes in AHRR methylation in lymphoblasts and pulmonary macrophages from smokers. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159, 141–151. [Google Scholar]
- Elliott, H.; Tillin, T.; McArdle, W.; Ho, K.; Duggirala, A.; Frayling, T.M.; Davey, S.G.; Hughes, A.D.; Chaturvedi, N.; Relton, C.L. Differences in smoking associated DNA methylation patterns in South Asians and Europeans. Clin. Epigenet. 2014, 6, 4. [Google Scholar] [CrossRef]
- Dogan, M.V.; Shields, B.; Cutrona, C.; Gao, L.; Gibbons, F.X.; Simons, R.L.; Monick, M.; Brody, G.H.; Tan, K.; Beach, S.R.H.; et al. The effect of smoking on DNA methylation of peripheral blood mononuclear cells from African American women. BMC Genom. 2014, 15, 151. [Google Scholar] [CrossRef] [PubMed]
- Zeilinger, S.; Kühnel, B.; Klopp, N.; Baurecht, H.; Kleinschmidt, A.; Gieger, C.; Weidinger, S.; Lattka, E.; Adamski, J.; Peters, A.; et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS ONE 2013, 8, e63812. [Google Scholar] [CrossRef] [PubMed]
- Philibert, R.; Beach, S.R.H.; Lei, M.-K.; Brody, G.H. Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking. Clin. Epigenet. 2013, 5, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Besingi, W.; Johansson, Å. Smoke related DNA methylation changes in the etiology of human disease. Hum. Mol. Genet. 2013, 23, 2290–2297. [Google Scholar] [CrossRef]
- Shenker, N.S.; Polidoro, S.; van Veldhoven, K.; Sacerdote, C.; Ricceri, F.; Birrell, M.A.; Belvisi, M.G.; Brown, R.; Vineis, P.; Flanagan, J.M. Epigenome-Wide association study in the European prospective investigation into cancer and nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum. Mol. Genet. 2012, 22, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Philibert, R.A.; Beach, S.R.H.; Brody, G.H. Demethylation of the aryl hydrocarbon receptor repressor as a biomarker for nascent smokers. Epigenetics 2012, 7, 1331–1338. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Horvath, S. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 2019, 11, 303–327. [Google Scholar] [CrossRef]
- Gorber, S.C.; Schofield-Hurwitz, S.; Hardt, J.; Levasseur, G.; Tremblay, M. The accuracy of self-reported smoking: A systematic review of the relationship between self-reported and cotinine-assessed smoking status. Nicotine Tobacco Res. 2009, 11, 12–24. [Google Scholar] [CrossRef]
- Caraballo, R.S.; Giovino, G.A.; Pechacek, T.F. Self-reported cigarette smoking vs. serum cotinine among U.S. adolescents. Nicotine Tobacco Res. 2004, 6, 19–25. [Google Scholar] [CrossRef]
- Fendrich, M.; Mackesy-Amiti, M.E.; Johnson, T.P.; Hubbell, A.; Wislar, J.S. Tobacco-reporting validity in an epidemiological drug-use survey. Addict. Behav. 2005, 30, 175–181. [Google Scholar] [CrossRef]
- Tsai, J.; Homa, D.M.; Gretzke, A.S.; Mahoney, M.; Sharapova, S.R.; Sosnoff, C.S.; Caron, K.T.; Wang, L.; Melstrom, R.C.; Trivers, K.F. Exposure to secondhand smoke among nonsmokers — United States, 1988–2014. Morbid. Mortal. Weekly Rep. 2018, 67, 1342–1346. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhang, Y.; Breitling, L.P.; Brenner, H. Relationship of tobacco smoking and smoking-related DNA methylation with epigenetic age acceleration. Oncotarget 2016, 7, 46878. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gao, X.; Just, A.C.; Colicino, E.; Wang, C.; Coull, B.A.; Hou, L.; Zheng, Y.; Vokonas, P.; Schwartz, J.; et al. Smoking-related DNA methylation is associated with DNA methylation phenotypic age acceleration: The veterans affairs normative aging study. Int. J. Environ. Res. Public Health 2019, 16, 2356. [Google Scholar] [CrossRef]
- Gao, X.; Jia, M.; Zhang, Y.; Breitling, L.P.; Brenner, H. DNA methylation changes of whole blood cells in response to active smoking exposure in adults: A systematic review of DNA methylation studies. Clin. Epigenet. 2015, 7, 113. [Google Scholar] [CrossRef]
- Zhang, Y.; Schöttker, B.; Florath, I.; Stock, C.; Butterbach, K.; Holleczek, B.; Mons, U.; Brenner, H. Smoking-associated DNA methylation biomarkers and their predictive value for all-cause and cardiovascular mortality. Environ. Health Perspect. 2015, 124, 67–74. [Google Scholar] [CrossRef]
- Zhang, Y.; Wilson, R.; Heiss, J.; Breitling, L.P.; Saum, K.U.; Schottker, B.; Holleczek, B.; Waldenberger, M.; Peters, A.; Brenner, H. DNA methylation signatures in peripheral blood strongly predict all-cause mortality. Nat. Commun. 2017, 8, 14617. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef]
- Zhao, W.; Ammous, F.; Ratliff, S.; Liu, J.; Yu, M.; Mosley, T.H.; Kardia, S.L.R.; Smith, J.A. Education and lifestyle factors are associated with DNA methylation clocks in older African Americans. Int. J. Environ. Res. Public Health 2019, 16, 3141. [Google Scholar] [CrossRef]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.G.; Lowe, R.; Adams, P.D.; Baccarelli, A.A.; Beck, S.; Bell, J.T.; Rakyan, V.K. DNA methylation aging clocks: Challenges and recommendations. Genome Biol. 2019, 20, 249. [Google Scholar] [CrossRef] [PubMed]
- Bocklandt, S.; Lin, W.; Sehl, M.E.; Sánchez, F.J.; Sinsheimer, J.S.; Horvath, S.; Vilain, E. Epigenetic predictor of age. PLoS ONE 2011, 6, e14821. [Google Scholar] [CrossRef] [PubMed]
- Field, A.E.; Robertson, N.A.; Wang, T.; Havas, A.; Ideker, T.; Adams, P.D. DNA methylation clocks in aging: Categories, causes, and consequences. Mol. Cell 2018, 71, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Jyhava, J.; Pedersen, N.; Hagg, S. Biological age predictors. EBioMedicine 2017, 21, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.T.; Seeboth, A.; Tsai, P.C.; Sun, D.; Quach, A.; Reiner, A.P.; Horvath, S. DNA methylation-based estimator of telomere length. Aging 2019, 11, 5895–5923. [Google Scholar] [CrossRef]
- Nordfjäll, K.; Svenson, U.; Norrback, K.; Adolfsson, R.; Roos, G. Large-scale parent–child comparison confirms a strong paternal influence on telomere length. Eur. J. Hum. Genet. 2010, 18, 385–389. [Google Scholar] [CrossRef]
- Verhulst, S.; Susser, E.; Factor-Litvak, P.R.; Simons, M.J.; Benetos, A.; Steenstrup, T.; Kark, J.D.; Aviv, A. Commentary: The reliability of telomere length measurements. Int. J. Epidemiol. 2015, 44, 1683–1686. [Google Scholar] [CrossRef]
- Andersen, A.M.; Lei, M.K.; Beach, S.H.R.; Philibert, R.A.; Sinha, S.; Colgan, J.D. Cigarette and cannabis smoking effects on GPR15+ helper T cell levels in peripheral blood: Relationships with epigenetic biomarkers. Genes 2020, 11, 149. [Google Scholar] [CrossRef]
- Bauer, M.; Linsel, G.; Fink, B.; Offenberg, K.; Hahn, A.M.; Sack, U.; Knaack, H.; Eszlinger, M.; Herberth, G. A varying T cell subtype explains apparent tobacco smoking induced single CpG hypomethylation in whole blood. Clin. Epigenet. 2015, 7, 81. [Google Scholar] [CrossRef]
- Gibbons, F.X.; Gerrard, M.; Cleveland, M.J.; Wills, T.A.; Brody, G.H. Perceived discrimination and substance use in African American parents and their children: A panel study. J. Personal. Soc. Psychol. 2004, 86, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Simons, R.L.; Lei, M.-K.; Beach, S.R.H.; Brody, G.H.; Philibert, R.A.; Gibbons, F.X. Social environment, genes, and aggression: Evidence supporting the differential susceptibility perspective. Am. Sociol. Rev. 2011, 76, 883–912. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, D.K.; Nurnberger, J.I., Jr. A rapid non-enzymatic method for the preparation of HMW DNA from blood for RFLP studies. Nucleic Acids Res. 1991, 19, 5444. [Google Scholar] [CrossRef] [PubMed]
- Philibert, R.A.; Terry, N.; Erwin, C.; Philibert, W.J.; Beach, S.R.H.; Brody, G.H. Methylation array data can simultaneously identify individuals and convey protected health information: An unrecognized ethical concern. Clin. Epigenet. 2014, 6, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.-K.; Simons, R.L.; Beach, S.R.; Philibert, R.A. Neighborhood disadvantage and biological aging: Using marginal structural models to assess the link between neighborhood census variables and epigenetic aging. J. Gerontol.: Series B 2019, 74, e50–e59. [Google Scholar] [CrossRef] [PubMed]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef] [PubMed]
- Hayes, A.F. Beyond Baron and Kenny: Statistical mediation analysis in the new millennioum. Commun. Monogr. 2009, 76, 408–420. [Google Scholar] [CrossRef]
- Muthén, L.K.; Muthén, B. Mplus User’s Guide. Eighth Edition; Muthén & Muthén: Los Angeles, CA, USA, 2017. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Self-Reported Cigarette Consumption | PACKYRS | cg05575921 | ||||
|---|---|---|---|---|---|---|
| r | p-Value | r | p-Value | r | p-Value | |
| Hannum | 0.046 | 0.304 | 0.057 | 0.208 | −0.008 | 0.851 |
| Horvath | −0.007 | 0.876 | −0.056 | 0.211 | 0.055 | 0.225 |
| PhenoAge | 0.122 ** | 0.007 | 0.133 ** | 0.003 | −0.109 * | 0.015 |
| mTL | −0.241 ** | 6.211 × 10 −8 | −0.261 ** | 3.917 × 10 −9 | 0.280 ** | 2.589 × 10 −10 |
| GrimAge | 0.650 ** | 1.409 × 10 −60 | 0.862 ** | 1.091 × 10 −146 | −0.778 ** | 5.480 × 10 −101 |
| ADM | 0.097 * | 0.032 | 0.163 ** | 2.910 × 10 −4 | −0.148 ** | 0.001 |
| BM2 | 0.052 | 0.253 | 0.007 | 0.875 | −0.031 | 0.496 |
| CystatinC | 0.099 * | 0.028 | 0.149 ** | 0.001 | −0.085 † | 0.059 |
| GDF15 | 0.222 ** | 6.319 × 10 −7 | 0.291 ** | 4.779 × 10 −11 | −0.265 ** | 2.219 × 10 −9 |
| Leptin | 0.001 | 0.997 | 0.006 | 0.901 | 0.033 | 0.460 |
| PAI1 | 0.087 † | 0.052 | 0.080 † | 0.077 | −0.055 | 0.220 |
| TIMP1 | 0.085 † | 0.060 | 0.063 | 0.162 | −0.046 | 0.307 |
| Self-Reported Cigarette Consumption | PACKYRS | cg05575921 | |||||
|---|---|---|---|---|---|---|---|
| β | p-Value | β | p-Value | β | p-Value | ||
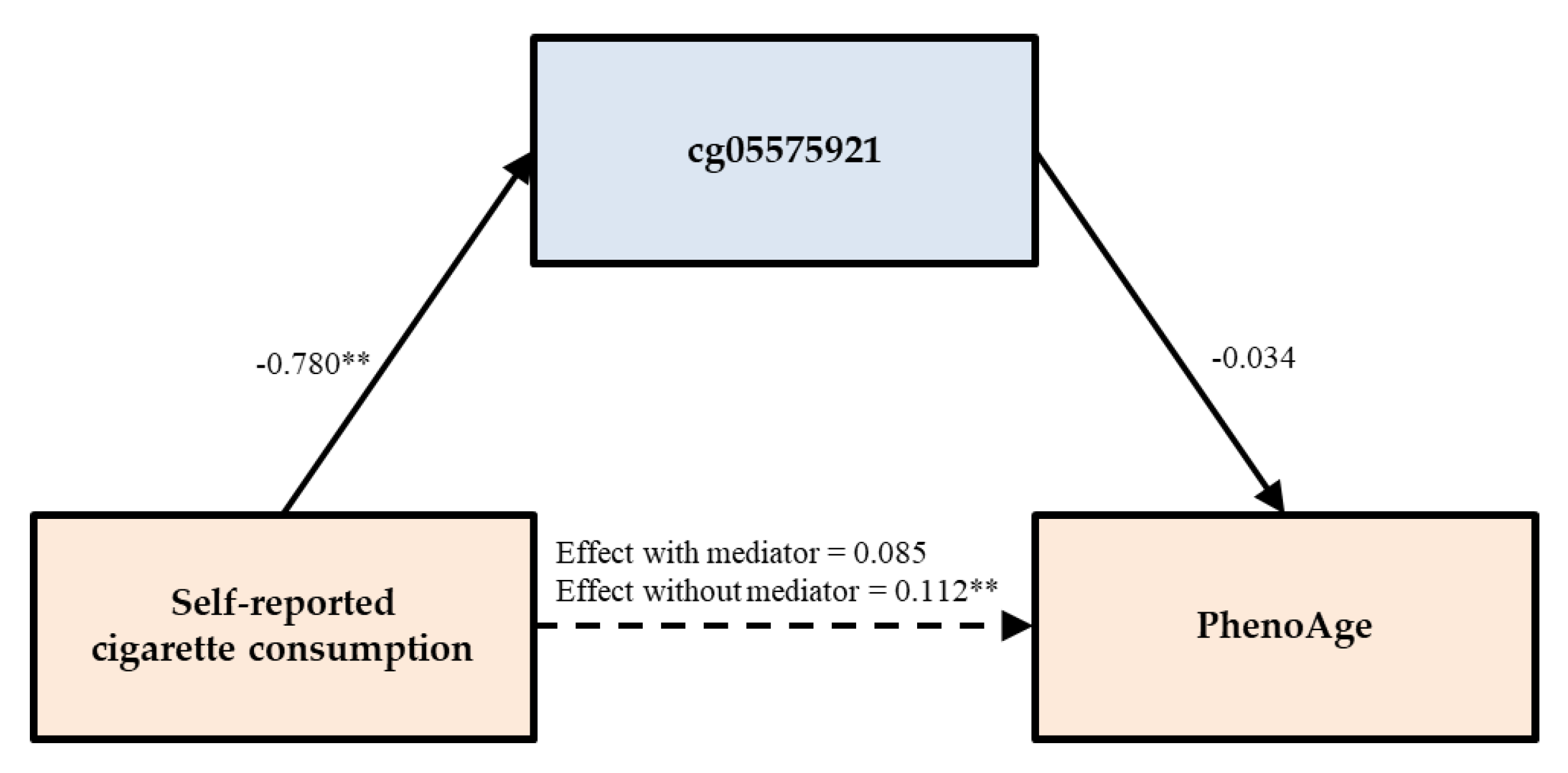
| PhenoAge | Model 1 | 0.111 ** | 0.007 | ||||
| Model 1A | 0.048 | 0.434 | 0.089 | 0.159 | |||
| Model 1B | 0.085 | 0.187 | −0.034 | 0.603 | |||
| mTL | Model 2 | −0.231 ** | 6.211 × 10 −8 | ||||
| Model 2A | −0.092 | 0.108 | −0.168 ** | 0.005 | |||
| Model 2B | −0.053 | 0.378 | 0.211 ** | 0.001 | |||
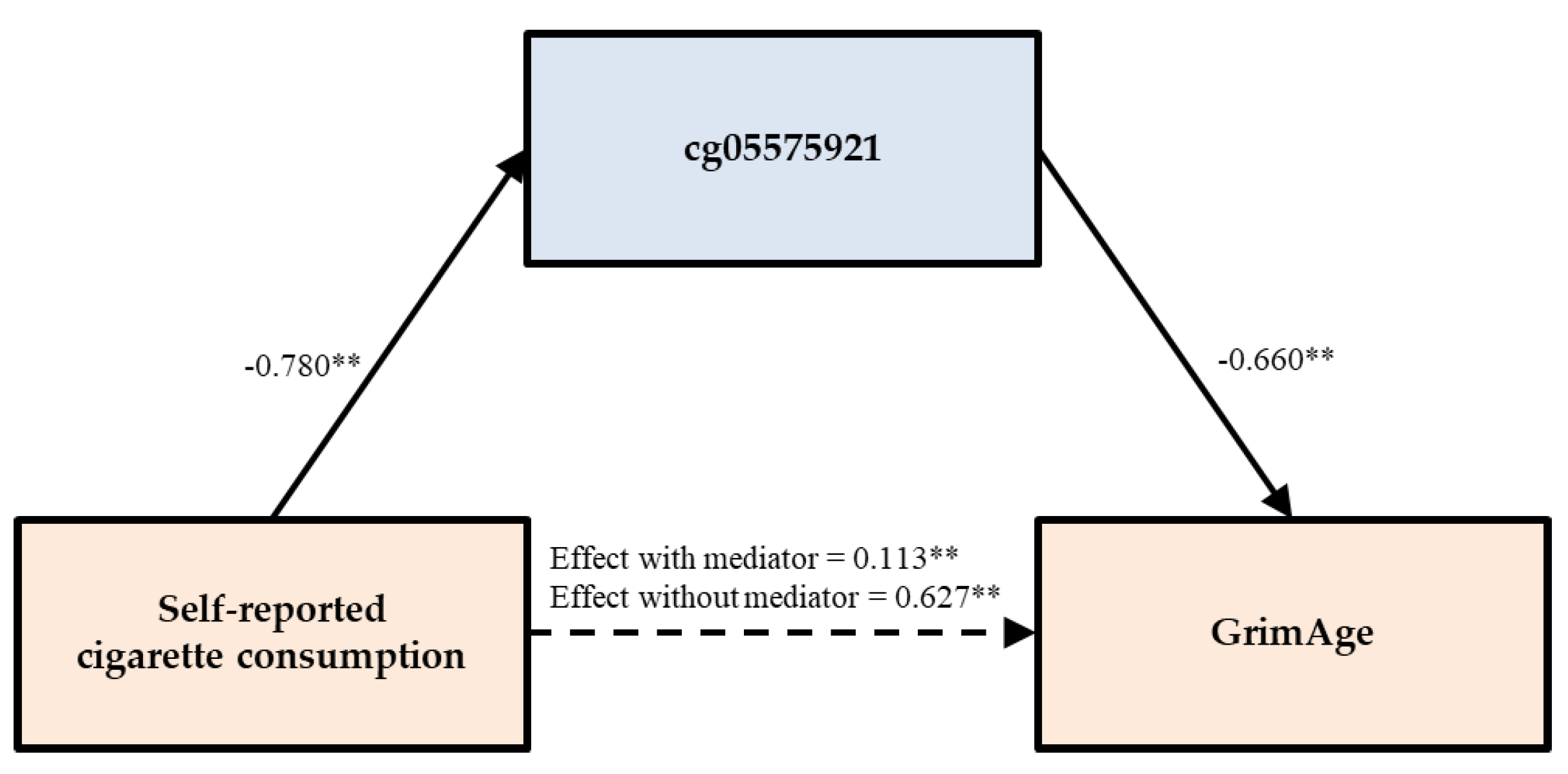
| GrimAge | Model 3 | 0.596 ** | 1.409 × 10 −60 | ||||
| Model 3A | 0.022 | 0.489 | 0.801 ** | 5.826 × 10 −88 | |||
| Model 3B | 0.110 ** | 0.007 | −0.644 ** | 9.494 × 10 −44 | |||
| ADM | Model 4 | 0.078 * | 0.032 | ||||
| Model 4A | −0.043 | 0.425 | 0.168 ** | 0.003 | |||
| Model 4B | −0.035 | 0.537 | −0.149 * | 0.011 | |||
| GDF15 | Model 5 | 0.224 ** | 6.319 × 10 −7 | ||||
| Model 5A | 0.014 | 0.825 | 0.292 ** | 1.700 × 10 −5 | |||
| Model 5B | 0.042 | 0.543 | −0.241 ** | 0.001 | |||
| Predictor | Mediators | Outcomes | Direct Effect | 95% CI | Indirect Effect | 95% CI | Mediation (%) |
|---|---|---|---|---|---|---|---|
| Self-reported smoking | PACKYRS | PhenoAge | 0.048 | (−0.097, 0.163) | 0.066 | (−0.038, 0.187) | -- |
| cg05575921 | 0.085 | (−0.050, 0.216) | 0.027 | (−0.090, 0.127) | -- | ||
| Self-reported smoking | PACKYRS | mTL | −0.093 | (−0.217, 0.031) | −0.126 * | (−0.224, −0.023) | 57.534 |
| cg05575921 | −0.054 | (−0.173, 0.070) | −0.166 * | (−0.264, −0.065) | 75.455 | ||
| Self-reported smoking | PACKYRS | GrimAge | 0.022 | (−0.040, 0.083) | 0.611 ** | (0.548, 0.668) | 96.524 |
| cg05575921 | 0.113 ** | (0.031, 0.192) | 0.514 ** | (0.448, 0.589) | 81.978 | ||
| Self-reported smoking | PACKYRS | ADM | −0.042 | (−0.140, 0.059) | 0.124 * | (0.043, 0.197) | 74.699 |
| cg05575921 | −0.035 | (−0.129, 0.071) | 0.116 * | (0.040, 0.198) | 76.821 | ||
| Self-reported smoking | PACKYRS | GDF15 | 0.014 | (−0.098, 0.141) | 0.218 ** | (0.106, 0.307) | 93.966 |
| cg05575921 | 0.042 | (−0.087, 0.170) | 0.188 ** | (0.071, 0.287) | 81.739 |
| Predictor | Mediators | Outcomes | Direct Effect | 95% CI | Indirect Effect | 95% CI | Mediation (%) |
|---|---|---|---|---|---|---|---|
| Self-reported smoking | PACKYRS cg05575921 | PhenoAge | −0.071 | (−0.233, 0.075) | 0.144 * | (0.011, 0.281) | 66.666 |
| −0.028 | (−0.190, 0.109) | 0.098 | (−0.021, 0.228) | -- | |||
| Self-reported smoking | PACKYRS cg05575921 | mTL | −0.010 | (−0.173, 0.135) | −0.161 * | (−0.287, −0.033) | 94.152 |
| 0.011 | (−0.130, 0.129) | −0.182 | (−0.293, −0.087) | 94.301 | |||
| Self-reported smoking | PACKYRS cg05575921 | GrimAge | −0.034 | (−0.122, 0.040) | 0.635 ** | (0.563, 0.712) | 94.918 |
| 0.038 | (−0.066, 0.133) | 0.557 ** | (0.480, 0.642) | 93.613 | |||
| Self-reported smoking | PACKYRS cg05575921 | ADM | −0.091 | (−0.199, 0.021) | 0.158 * | (0.072, 0.238) | 63.454 |
| −0.091 | (−0.200, 0.025) | 0.157 * | (0.070, 0.258) | 63.306 | |||
| Self-reported smoking | PACKYRS cg05575921 | GDF15 | −0.029 | (−0.147, 0.098) | 0.240 ** | (0.123, 0.329) | 89.219 |
| −0.004 | (−0.136, 0.132) | 0.213 ** | (0.094, 0.316) | 98.157 |
| Predictor | Mediators | Outcomes | Direct effect | 95% CI | Indirect effect | 95% CI | Mediation (%) |
|---|---|---|---|---|---|---|---|
| Self-reported smoking | PACKYRS cg05575921 | PhenoAge | 0.031 | (−0.123, 0.159) | 0.087 | (−0.028, 0.223) | -- |
| 0.081 | (−0.082, 0.218) | 0.034 | (−0.104, 0.167) | -- | |||
| Self-reported smoking | PACKYRS cg05575921 | mTL | −0.088 | (−0.214, 0.043) | −0.137 * | (−0.256, −0.039) | 60.889 |
| −0.055 | (−0.196, 0.068) | −0.170 * | (−0.278, −0.066) | 75.556 | |||
| Self-reported smoking | PACKYRS cg05575921 | GrimAge | 0.029 | (−0.040, 0.094) | 0.616 ** | (0.548, 0.683) | 95.504 |
| 0.106 ** | (0.019, 0.192) | 0.536 ** | (0.460, 0.617) | 83.489 | |||
| Self-reported smoking | PACKYRS cg05575921 | ADM | −0.046 | (−0.169, 0.068) | 0.131 * | (0.059, 0.222) | 74.011 |
| −0.045 | (−0.164, 0.061) | 0.130 * | (0.041, 0.228) | 74.286 | |||
| Self-reported smoking | PACKYRS cg05575921 | GDF15 | 0.015 | (−0.115, 0.148) | 0.220 ** | (0.110, 0.320) | 93.617 |
| 0.036 | (−0.110, 0.176) | 0.197 ** | (0.087, 0.310) | 84.549 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, M.-K.; Gibbons, F.X.; Simons, R.L.; Philibert, R.A.; Beach, S.R.H. The Effect of Tobacco Smoking Differs across Indices of DNA Methylation-Based Aging in an African American Sample: DNA Methylation-Based Indices of Smoking Capture These Effects. Genes 2020, 11, 311. https://doi.org/10.3390/genes11030311
Lei M-K, Gibbons FX, Simons RL, Philibert RA, Beach SRH. The Effect of Tobacco Smoking Differs across Indices of DNA Methylation-Based Aging in an African American Sample: DNA Methylation-Based Indices of Smoking Capture These Effects. Genes. 2020; 11(3):311. https://doi.org/10.3390/genes11030311
Chicago/Turabian StyleLei, Man-Kit, Frederick X. Gibbons, Ronald L. Simons, Robert A. Philibert, and Steven R. H. Beach. 2020. "The Effect of Tobacco Smoking Differs across Indices of DNA Methylation-Based Aging in an African American Sample: DNA Methylation-Based Indices of Smoking Capture These Effects" Genes 11, no. 3: 311. https://doi.org/10.3390/genes11030311
APA StyleLei, M.-K., Gibbons, F. X., Simons, R. L., Philibert, R. A., & Beach, S. R. H. (2020). The Effect of Tobacco Smoking Differs across Indices of DNA Methylation-Based Aging in an African American Sample: DNA Methylation-Based Indices of Smoking Capture These Effects. Genes, 11(3), 311. https://doi.org/10.3390/genes11030311