mRNAsi Index: Machine Learning in Mining Lung Adenocarcinoma Stem Cell Biomarkers

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Software and R Packages
2.2. Database and mRNAsi Index
2.3. Differentially Expressed Genes (DEGs) Analysis
2.4. Overall Survival Curve
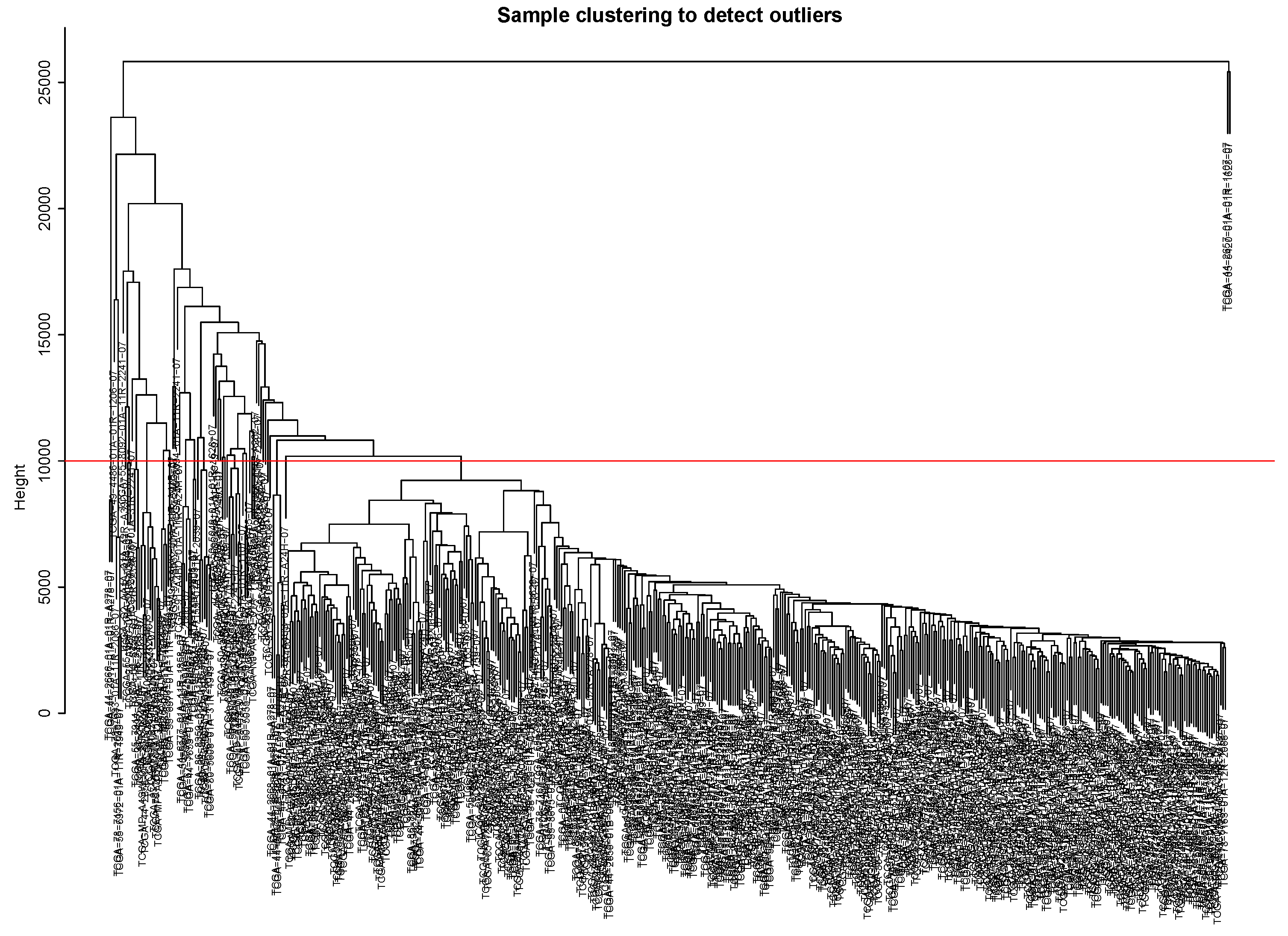
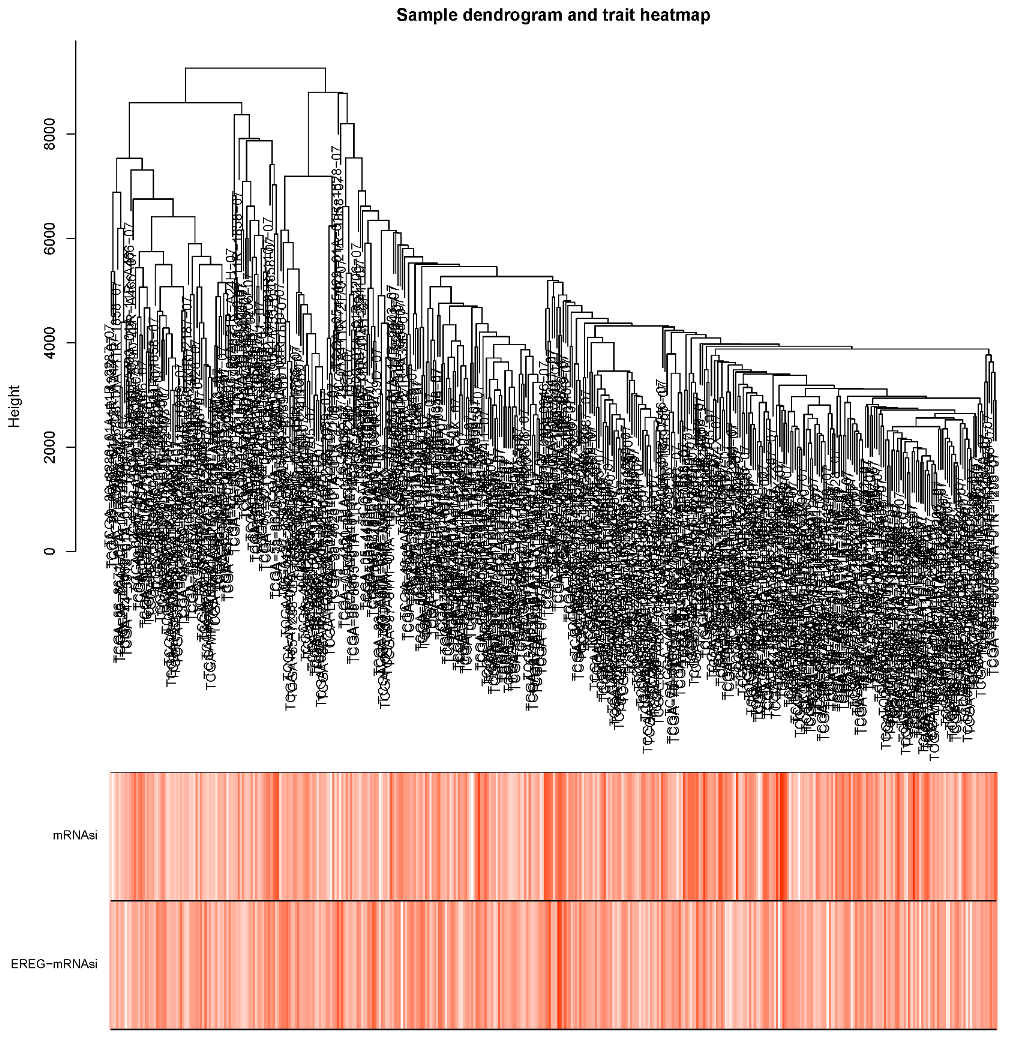
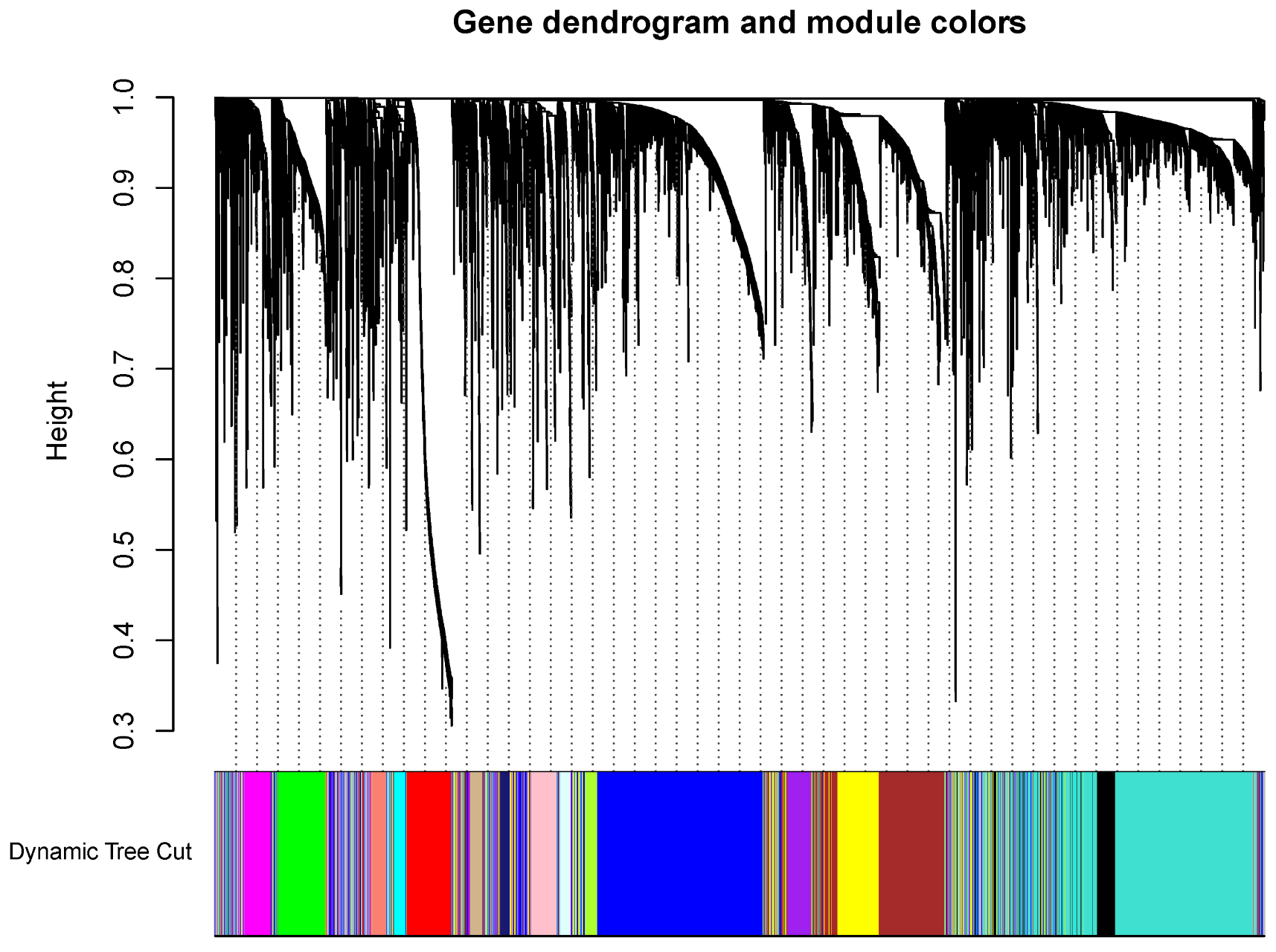
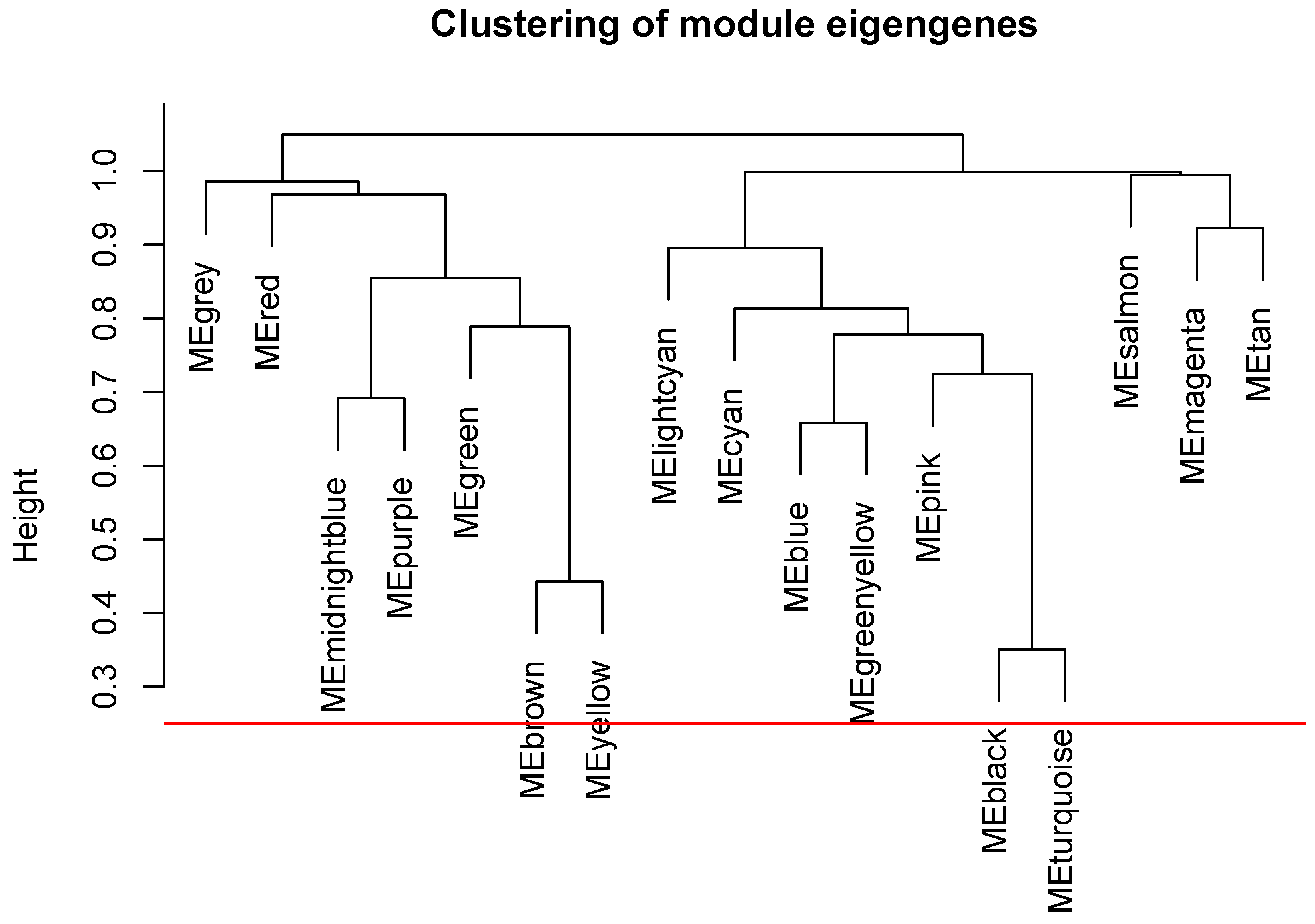
2.5. The WGCNA Analysis to Filter Key Genes
2.6. Gene Co-Expression Analysis
2.7. Construction of Protein–Protein Interaction (PPI) Network
2.8. Enrichment Analysis of DEGs
2.9. Venn Diagrams
3. Results
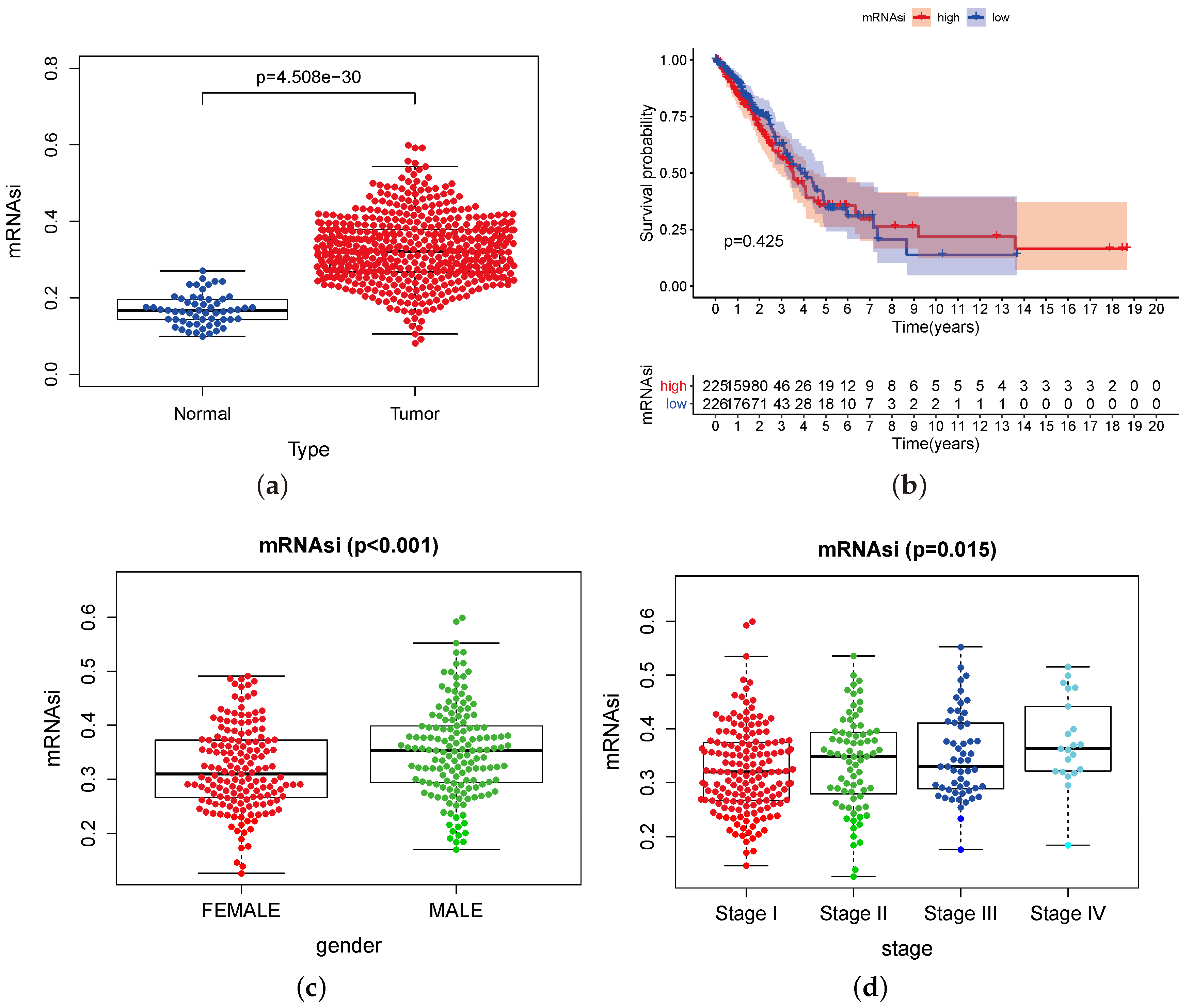
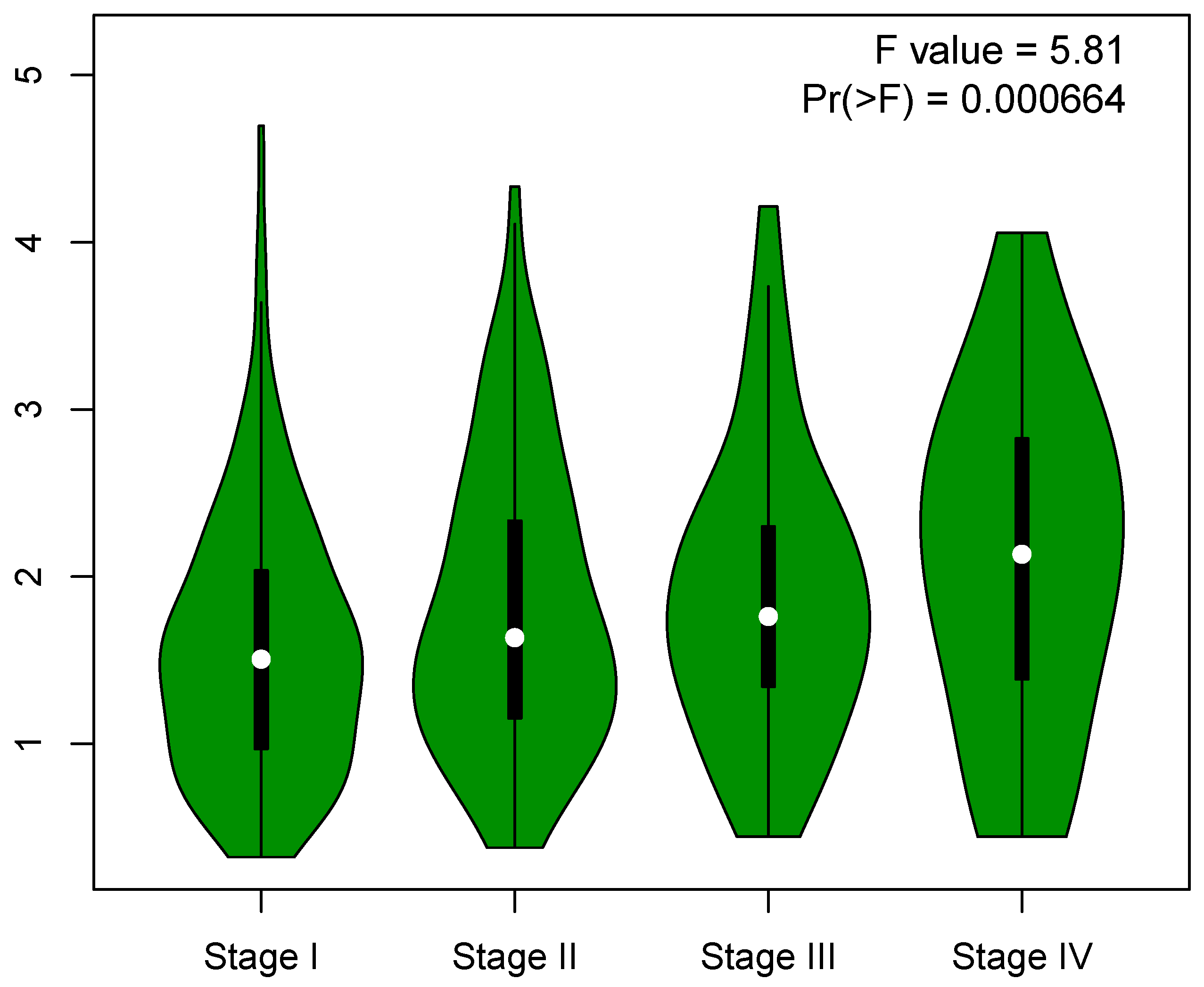
3.1. mRNAsi Index Is Significantly Associated with Lung Adenocarcinoma
3.2. The Screening of DEGs and Construction of the WGCNA Co-Expression Network
3.3. Extracting and Profile of the Key Genes
3.4. Protein–Protein Interactions (PPI) among Genes of Each Module
3.5. Functional Enrichment Analysis of Key Genes
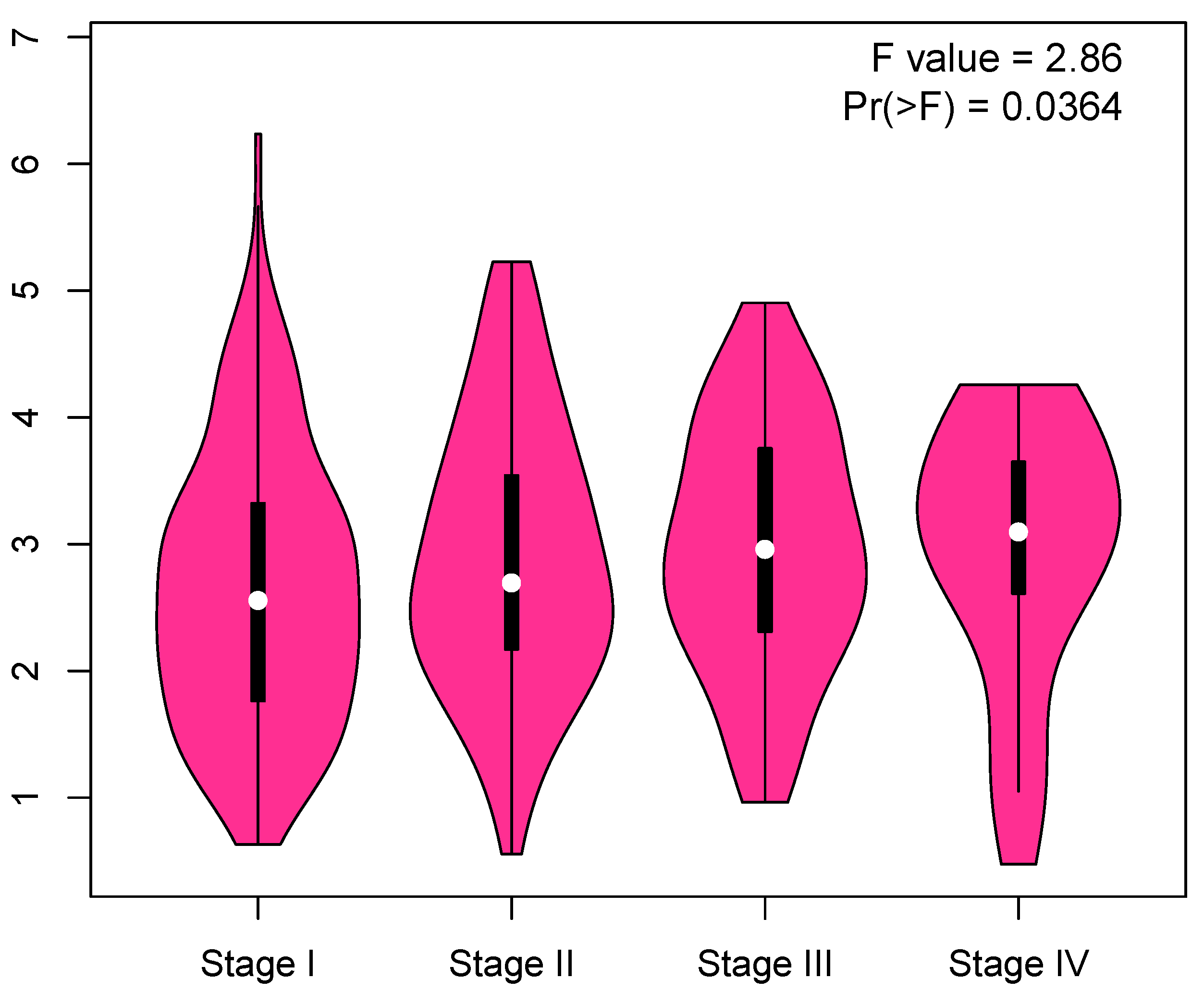
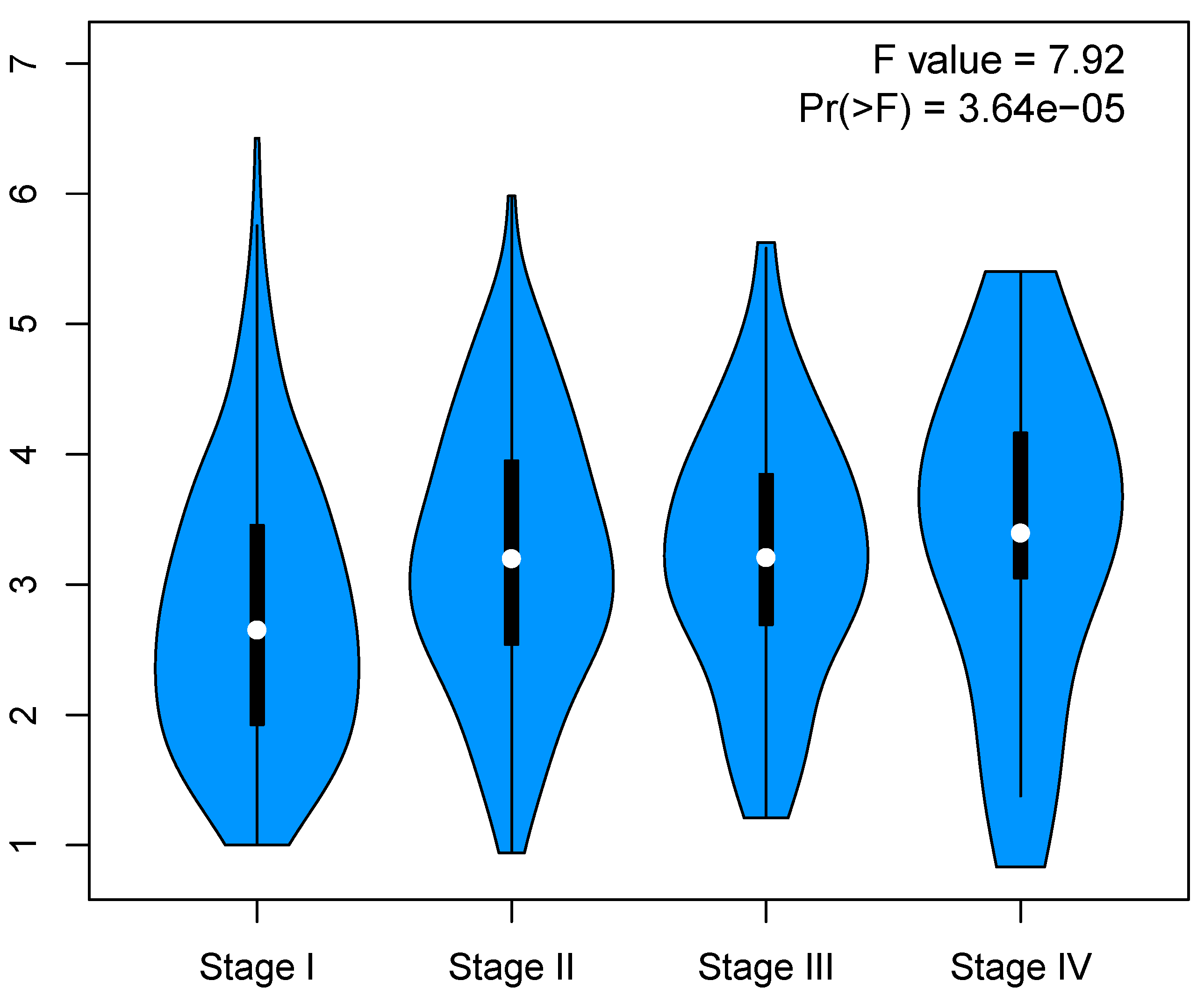
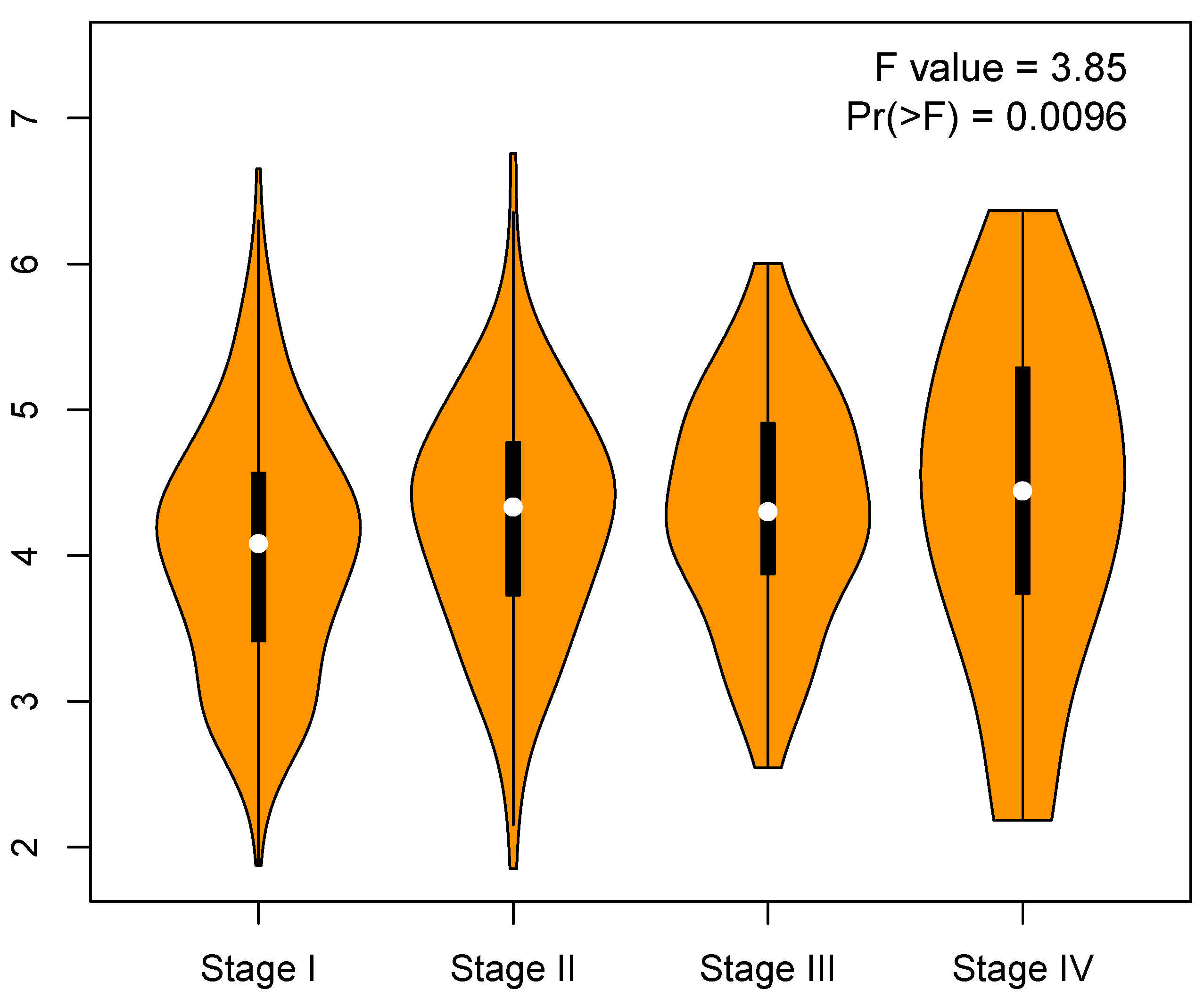
3.6. Validation of Key Genes
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. The Steps to Draw the Dendrograms





Appendix B. Correlation between Cell Cycle Key Gene Expression and Tumor Stages in LUAD Patients













References
- Tsolou, A.; Lamprou, I.; Fortosi, A.O.; Liousia, M.; Giatromanolaki, A.; Koukourakis, M.I. ‘Stemness’ and ‘senescence’ related escape pathways are dose dependent in lung cancer cells surviving post irradiation. Life Sci. 2019, 232, 116562. [Google Scholar] [CrossRef] [PubMed]
- Leon, G.; MacDonagh, L.; Finn, S.P.; Cuffe, S.; Barr, M.P. Cancer stem cells in drug resistant lung cancer: Targeting cell surface markers and signaling pathways. Pharmacol. Therap. 2016, 158, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Wang, M.; Li, X.; Wu, J.; Chen, S.; Du, N.; Li, K.; Wang, J.; Xu, C.; Ren, H.; et al. TUSC7 suppression of Notch activation through sponging MiR-146 recapitulated the asymmetric cell division in lung adenocarcinoma stem cells. Life Sci. 2019, 232, 116630. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Dong, D.; Zhang, T.; Xu, W. Long non-coding RNA FENDRR attenuates the stemness of non-small cell lung cancer cells via decreasing multidrug resistance gene 1 (MDR1) expression through competitively binding with RNA binding protein HuR. Eur. J. Pharmacol. 2019, 853, 345–352. [Google Scholar] [CrossRef]
- Liu, Q.; Bai, W.; Huang, F.; Tang, J.; Lin, X. Downregulation of microRNA-196a inhibits stem cell self-renewal ability and stemness in non-small-cell lung cancer through upregulating GPX3 expression. Int. J. Biochem. Cell Biol. 2019, 115, 105571. [Google Scholar] [CrossRef] [PubMed]
- Zakaria, N.; Yusoff, N.M.; Zakaria, Z.; Lim, M.N.; Baharuddin, P.J.; Fakiruddin, K.S.; Yahaya, B. Human non-small cell lung cancer expresses putative cancer stem cell markers and exhibits the transcriptomic profile of multipotent cells. BMC Cancer 2015, 15, 84. [Google Scholar] [CrossRef]
- Miyata, T.; Yoshimatsu, T.; So, T.; Oyama, T.; Uramoto, H.; Osaki, T.; Nakanishi, R.; Tanaka, F.; Nagaya, H.; Gotoh, A. Cancer stem cell markers in lung cancer. Personal. Med. Univ. 2015, 4, 40–45. [Google Scholar] [CrossRef]
- Tanner, N.T.; Thomas, N.A.; Ward, R.; Rojewski, A.; Gebregziabher, M.; Toll, B.; Silvestri, G.A. Association of Cigarette Type With Lung Cancer Incidence and Mortality: Secondary Analysis of the National Lung Screening Trial. JAMA Intern. Med. 2019, 3487, 1710–1712. [Google Scholar] [CrossRef]
- Horikoshi, Y.; Yan, Y.; Terashvili, M. Fatty Acid-Treated Induced Pluripotent Stem Cell-Derived Human Cardiomyocytes Exhibit Adult Cardiomyocyte-Like Energy Metabolism Phenotypes. Cells 2019, 8, 1095. [Google Scholar] [CrossRef]
- Paolillo, M.; Colombo, R.; Serra, M.; Belvisi, L.; Papetti, A.; Ciusani, E.; Comincini, S.; Schinelli, S. Stem-like Cancer Cells in a Dynamic 3D Culture System: A Model to Study Metastatic Cell Adhesion and Anti-cancer Drugs. Cells 2019, 8, 1434. [Google Scholar] [CrossRef]
- MacDonagh, L.; Gray, S.G.; Breen, E.; Cuffe, S.; Finn, S.P.; O’Byrne, K.J.; Barr, M.P. Lung cancer stem cells: The root of resistance. Cancer Lett. 2016, 372, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Sterlacci, W.; Savic, S.; Fiegl, M.; Obermann, E.; Tzankov, A. Putative Stem Cell Markers in Non–Small-Cell Lung Cancer: A Clinicopathologic Characterization. J. Thor. Oncol. 2014, 9, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Logan, S.; Arzua, T.; Canfield, S.G.; Seminary, E.R.; Sison, S.L.; Ebert, A.D.; Bai, X. Studying Human Neurological Disorders Using Induced Pluripotent Stem Cells: From 2D Monolayer to 3D Organoid and Blood Brain Barrier Models. Comprehens. Physiol. 2019, 9, 565–611. [Google Scholar]
- Julian, W.; Thomas, D. Minocycline impairs TNF-α-induced cell fusion of M13SV1-Cre cells with MDA-MB-435-pFDR1 cells by suppressing NF-κB transcriptional activity and its induction of target-gene expression of fusion-relevant factors. Cell Commun. Signal. 2019, 17, 71. [Google Scholar]
- Piperi, C.; Papavassiliou, K.A.; Papavassiliou, A.G. Pivotal Role of STAT3 in Shaping Glioblastoma Immune Microenvironment. Cells 2019, 8, 1398. [Google Scholar] [CrossRef]
- Mario, C.; Giuseppe, L.; Stefano, I.; Vittorina, Z. PD-1/PD-L1 immune-checkpoint inhibitors in glioblastoma: A concise review. Crit. Rev. Oncol./Hematol. 2019, 135, 128–134. [Google Scholar]
- Bonanno, L.; Zulato, E.; Pavan, A.; Attili, I.; Pasello, G.; Conte, P.; Indraccolo, S. LKB1 and Tumor Metabolism: The Interplay of Immune and Angiogenic Microenvironment in Lung Cancer. Int. J. Mol. Sci. 2019, 20, 1874. [Google Scholar] [CrossRef]
- Kim, R.; Keam, B.; Kim, S.; Kim, M.; Kim, S.H.; Kim, J.W.; Kim, Y.J.; Kim, T.M.; Jeon, Y.K.; Kim, D.W.; et al. Differences in tumor microenvironments between primary lung tumors and brain metastases in lung cancer patients: Therapeutic implications for immune checkpoint inhibitors. BMC Cancer 2019, 19, 19. [Google Scholar] [CrossRef]
- Sun, Y.; Peng, M.; Zhou, Y.; Huang, Y.; Mao, S. Application of Machine Learning in Wireless Networks: Key Techniques and Open Issues. IEEE Commun. Surv. Tutor. 2019. [Google Scholar] [CrossRef]
- Abdi, H.; Williams, L.J. Principal component analysis. Wiley Interdiscip. Rev. Comput. Stat. 2010, 2, 433–459. [Google Scholar] [CrossRef]
- Sokolov, A.; Paull, E.O.; Stuart, J.M. One-class detection of cell states in tumor subtypes. Pac. Symp. Biocomput. 2016, 21, 405–416. [Google Scholar] [PubMed]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kamińska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Lánczky, A.; Menyhárt, O.; Győrffy, B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep. 2018, 8, 9227. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Vasaikar, S.; Straub, P.; Wang, J.; Zhang, B. LinkedOmics: Analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018, 46, D956–D963. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, G.; Tian, H.; Hu, Y.; Wu, S.; Geng, Y.; Lin, K.; Wu, W. Sulforaphane metabolites cause apoptosis via microtubule disruption in cancer. Endocrine-Relat. Cancer 2018, 25, 255–268. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, Y.; Zheng, Z.; Li, J.; Yan, Y.; Wu, W. Sulforaphane metabolites reduce resistance to paclitaxel via microtubule disruption. Cell Death Disease 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kustatscher, G.; Grabowski, P.; Schrader, T.A.; Passmore, J.B.; Schrader, M.; Rappsilber, J. Co-regulation map of the human proteome enables identification of protein functions. Nat. Biotechnol. 2019, 37, 1361–1371. [Google Scholar] [CrossRef]
- Sun, Y.; Zheng, S.; Torossian, A.; Speirs, C.K.; Schleicher, S.; Giacalone, N.J.; Carbone, D.P.; Zhao, Z.; Lu, B. Role of insulin-like growth factor-1 signaling pathway in cisplatin-resistant lung cancer cells. Int. J. Radiat. Oncol. Biol. Phys. 2012, 82, e563–e572. [Google Scholar] [CrossRef] [PubMed]
- Colombino, M.; Paliogiannis, P.; Cossu, A.; Santeufemia, D.A.; Sini, M.C.; Casula, M.; Palomba, G.; Manca, A.; Pisano, M.; Doneddu, V.; et al. EGFR, KRAS, BRAF, ALK, and cMET genetic alterations in 1440 Sardinian patients with lung adenocarcinoma. BMC Pulm Med. 2019, 19, 209. [Google Scholar] [CrossRef] [PubMed]
- Karatrasoglou, E.A.; Chatziandreou, I.; Sakellariou, S.; Stamopoulos, K.; Kavantzas, N.; Lazaris, A.C.; Korkolopoulou, P.; Saetta, A.A. Association between PD-L1 expression and driver gene mutations in non-small cell lung cancer patients: Correlation with clinical data. Virchows Arch. 2020. [Google Scholar] [CrossRef]
- Choi, H.; Paeng, J.C.; Kim, D.W.; Lee, J.K.; Park, C.M.; Kang, K.W.; Chung, J.K.; Lee, D.S. Metabolic and metastatic characteristics of ALK-rearranged lung adenocarcinoma on FDG PET/CT. Lung Cancer 2013, 79, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Ricci, F.; Brunelli, L.; Affatato, R.; Chilà, R.; Verza, M.; Indraccolo, S.; Falcetta, F.; Fratelli, M.; Fruscio, R.; Pastorelli, R.; et al. Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts. Ther. Adv. Med. Oncol. 2018, 11. [Google Scholar] [CrossRef]
- Choi, H.S.; Kim, J.-H.; Kim, S.-L.; Lee, D.-S. Disruption of the NF-κB/IL-8 Signaling Axis by Sulconazole Inhibits Human Breast Cancer Stem Cell Formation. Cells 2019, 8, 1007. [Google Scholar] [CrossRef]
- Julian, W.; Thomas, D. Cell Fusion in Human Cancer: The Dark Matter Hypothesis. Cells 2019, 8, 132. [Google Scholar]
- Wang, Y.; Zhou, L.; Qing, Q.; Li, Y.; Li, L.; Dong, X.; Xiao, B. Gene expression profile of cancer stem like cells in the SW480 colon adenocarcinoma cell line. Oncol. Rep. 2019, 42, 386–398. [Google Scholar] [CrossRef]
- Han, J.Y.; Han, Y.K.; Park, G.Y.; Kim, S.D.; Lee, C.G. Bub1 is required for maintaining cancer stem cells in breast cancer cell lines. Sci. Rep. 2015, 5, 15993. [Google Scholar] [CrossRef]
- Su, J.; Zhu, D.; Huo, Z.; Gingold, J.A.; Ang, Y.S.; Tu, J.; Zhou, R.; Lin, Y.; Luo, H.; Yang, H.; et al. Genomic Integrity Safeguards Self-Renewal in Embryonic Stem Cells. Cell Rep. 2019, 28, 1400–1409. [Google Scholar] [CrossRef]
- Han, T.; Yin, Q.; Wan, L. Cycling for renewal: Cell cycle machinery maintains prostate cancer stem-like cells. EBioMedicine 2019, 42, 24–25. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Hu, G.; Wang, H.; Li, Z.; Guo, Z. PLK1 inhibitor facilitates the suppressing effect of temozolomide on human brain glioma stem cells. J. Cell. Mol. Med. 2018, 22, 5300–5310. [Google Scholar] [CrossRef] [PubMed]
- Hammoud, L.; Adams, J.R.; Loch, A.J.; Marcellus, R.C.; Uehling, D.E.; Aman, A.; Fladd, C.; McKee, T.D.; Jo, C.E.; Al-Awar, R.; et al. Identification of RSK and TTK as Modulators of Blood Vessel Morphogenesis Using an Embryonic Stem Cell-Based Vascular Differentiation Assay. Stem Cell Rep. 2016, 7, 787–801. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, J.; Xie, Y.; Bai, X.; Wang, N.; Yu, H.; Deng, Z.; Lian, M.; Yu, S.; Liu, H.; Xie, W.; et al. Targeting dual specificity protein kinase TTK attenuates tumorigenesis of glioblastoma. Oncotarget 2019, 9, 3081. [Google Scholar] [CrossRef] [PubMed]
- Abe, S.; Yamamoto, K.; Kurata, M.; Abe-Suzuki, S.; Horii, R.; Akiyama, F.; Kitagawa, M. Targeting MCM2 function as a novel strategy for the treatment of highly malignant breast tumors. Oncotarget 2015, 6, 34892. [Google Scholar] [CrossRef]
- Alcaraz-Sanabria, A.; Nieto-Jiménez, C.; Corrales-Sánchez, V.; Serrano-Oviedo, L.; Andrés-Pretel, F.; Montero, J.C.; Burgos, M.; Llopis, J.; Galán-Moya, E.M.; Pandiella, A.; et al. Synthetic Lethality Interaction Between Aurora Kinases and CHEK1 Inhibitors in Ovarian Cancer. Mol. Cancer Therap. 2017, 16, 2552–2562. [Google Scholar] [CrossRef]
- Gong, D.; Feng, P.C.; Ke, X.F.; Kuang, H.L.; Pan, L.L.; Ye, Q.; Wu, J.B. Silencing Long Non-coding RNA LINC01224 Inhibits Hepatocellular Carcinoma Progression via MicroRNA-330-5p-Induced Inhibition of CHEK1. Mol. Ther. Nucleic Acids 2020, 19, 482–497. [Google Scholar] [CrossRef]
- Han, Y.G.; Yun, M.; Choi, M.; Lee, S.G.; Kim, H. TRAIP regulates Histone H2B monoubiquitination in DNA damage response pathways. Oncol. Rep. 2019, 41, 3305–3312. [Google Scholar] [CrossRef]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Tseng, J.T.-C.; Lien, I.-C.; Li, F.; Wu, W.; Li, H. mRNAsi Index: Machine Learning in Mining Lung Adenocarcinoma Stem Cell Biomarkers. Genes 2020, 11, 257. https://doi.org/10.3390/genes11030257
Zhang Y, Tseng JT-C, Lien I-C, Li F, Wu W, Li H. mRNAsi Index: Machine Learning in Mining Lung Adenocarcinoma Stem Cell Biomarkers. Genes. 2020; 11(3):257. https://doi.org/10.3390/genes11030257
Chicago/Turabian StyleZhang, Yitong, Joseph Ta-Chien Tseng, I-Chia Lien, Fenglan Li, Wei Wu, and Hui Li. 2020. "mRNAsi Index: Machine Learning in Mining Lung Adenocarcinoma Stem Cell Biomarkers" Genes 11, no. 3: 257. https://doi.org/10.3390/genes11030257
APA StyleZhang, Y., Tseng, J. T.-C., Lien, I.-C., Li, F., Wu, W., & Li, H. (2020). mRNAsi Index: Machine Learning in Mining Lung Adenocarcinoma Stem Cell Biomarkers. Genes, 11(3), 257. https://doi.org/10.3390/genes11030257