Whole Exome Sequence Analysis Provides Novel Insights into the Genetic Framework of Childhood-Onset Pulmonary Arterial Hypertension

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Cohort and Clinical Assessment
2.2. Exome Sequencing
2.3. Candidate Gene Analysis
3. Results
3.1. WES Analysis of the cPAH Cohort
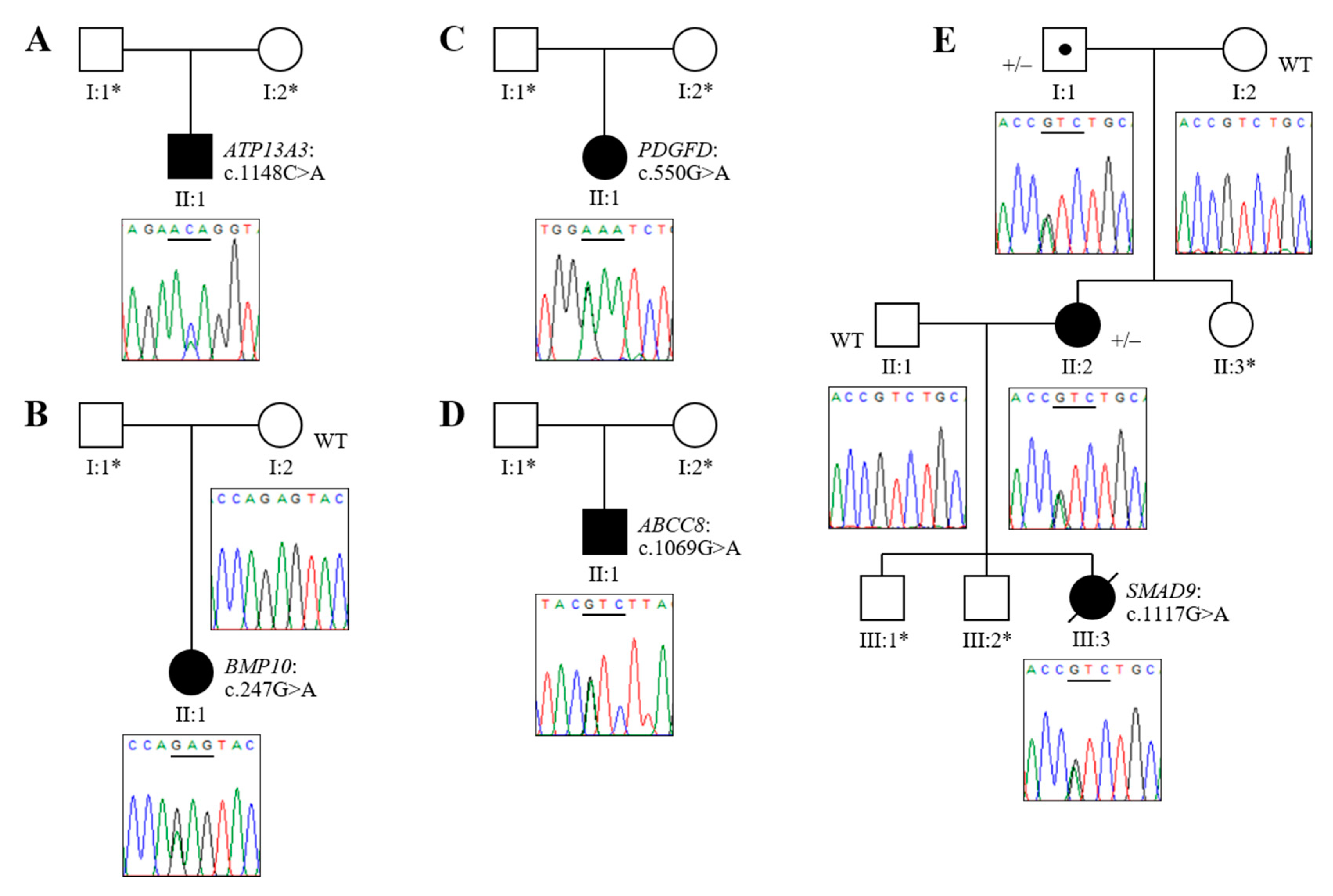
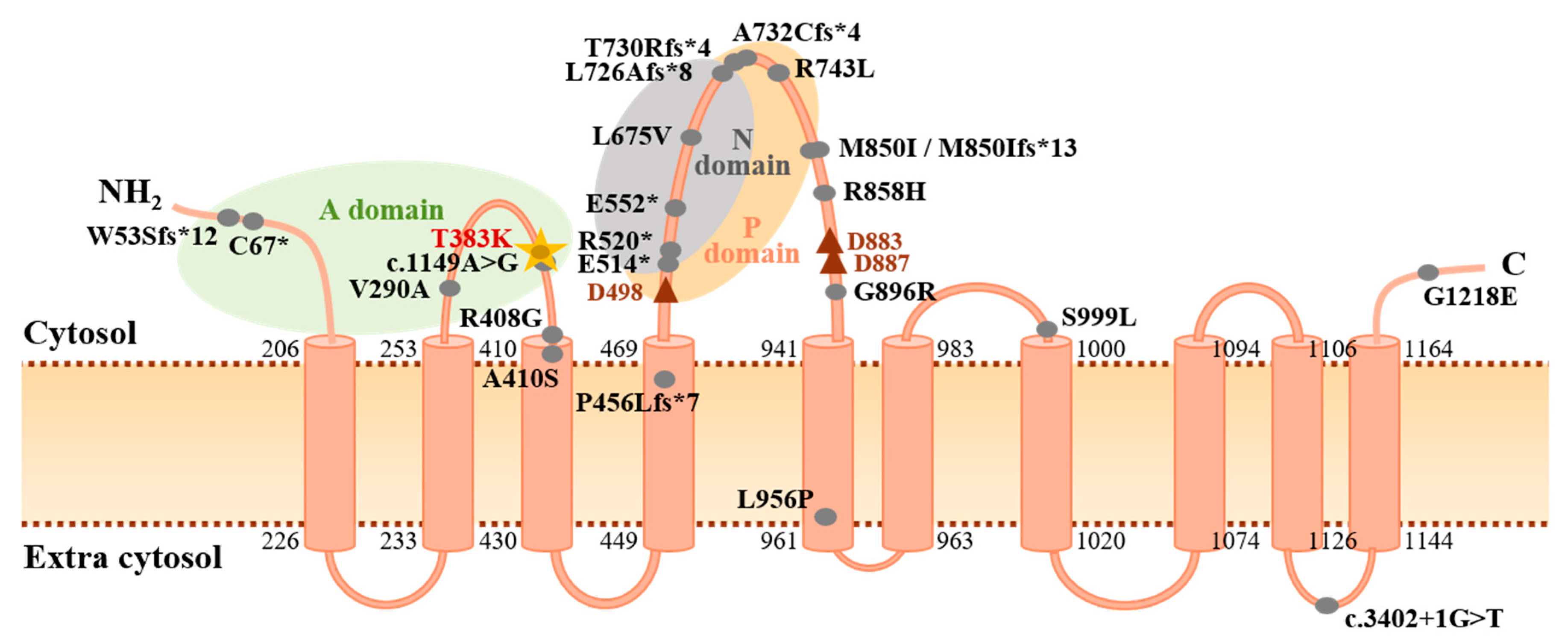
3.2. Novel Association of ATP13A3 with Paediatric APAH-CHD
3.3. Novel Mutations of BMP10, PDGFD and ABCC8 in Childhood IPAH
3.4. Analysis of HPAH Samples Identifies a Potential Molecular Defect of SMAD9
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Machado, R.D.; Eickelberg, O.; Elliott, C.G.; Geraci, M.W.; Hanaoka, M.; Loyd, J.E.; Newman, J.H.; Phillips, J.A.; Soubrier, F.; Trembath, R.C.; et al. Genetics and genomics of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2009, 54, S32–S42. [Google Scholar] [CrossRef]
- Machado, R.D.; Aldred, M.A.; James, V.; Harrison, R.E.; Patel, B.; Schwalbe, E.C.; Gruenig, E.; Janssen, B.; Koehler, R.; Seeger, W.; et al. Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum. Mutat. 2006, 27, 121–132. [Google Scholar] [CrossRef]
- O’Callaghan, D.S.; Humbert, M. A critical analysis of survival in pulmonary arterial hypertension. Eur. Respir. Rev. 2012, 21, 218–222. [Google Scholar] [CrossRef]
- van Loon, R.L.E.; Roofthooft, M.T.R.; Hillege, H.L.; ten Harkel, A.D.J.; van Osch-Gevers, M.; Delhaas, T.; Kapusta, L.; Strengers, J.L.M.; Rammeloo, L.; Clur, S.-A.B.; et al. Pediatric pulmonary hypertension in the Netherlands: Epidemiology and characterization during the period 1991 to 2005. Circulation 2011, 124, 1755–1764. [Google Scholar] [CrossRef]
- Li, L.; Jick, S.; Breitenstein, S.; Hernandez, G.; Michel, A.; Vizcaya, D. Pulmonary arterial hypertension in the USA: An epidemiological study in a large insured pediatric population. Pulm. Circ. 2017, 7, 126–136. [Google Scholar] [CrossRef]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaici, A.; Weitzenblum, E.; Cordier, J.-F.; Chabot, F.; et al. Pulmonary arterial hypertension in France: Results from a national registry. Am. J. Respir. Crit. Care Med. 2006, 173, 1023–1030. [Google Scholar] [CrossRef]
- Ivy, D. Pulmonary hypertension in children. Cardiol. Clin. 2016, 34, 451–472. [Google Scholar] [CrossRef]
- Zhu, N.; Gonzaga-Jauregui, C.; Welch, C.L.; Ma, L.; Qi, H.; King, A.K.; Krishnan, U.; Rosenzweig, E.B.; Ivy, D.D.; Austin, E.D.; et al. Exome sequencing in children with pulmonary arterial hypertension demonstrates differences compared with adults. Circ. Genom. Precis. Med. 2018, 11, e001887. [Google Scholar] [CrossRef]
- Chida, A.; Shintani, M.; Matsushita, Y.; Sato, H.; Eitoku, T.; Nakayama, T.; Furutani, Y.; Hayama, E.; Kawamura, Y.; Inai, K.; et al. Mutations of NOTCH3 in childhood pulmonary arterial hypertension. Mol. Genet. Genom. Med. 2014, 2, 229–239. [Google Scholar] [CrossRef]
- Gräf, S.; Haimel, M.; Bleda, M.; Hadinnapola, C.; Southgate, L.; Li, W.; Hodgson, J.; Liu, B.; Salmon, R.M.; Southwood, M.; et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat. Commun. 2018, 9, 1416. [Google Scholar] [CrossRef]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; Loyd, J.E. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801899. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Pauciulo, M.W.; Welch, C.L.; Lutz, K.A.; Coleman, A.W.; Gonzaga-Jauregui, C.; Wang, J.; Grimes, J.M.; Martin, L.J.; He, H.; et al. Novel risk genes and mechanisms implicated by exome sequencing of 2572 individuals with pulmonary arterial hypertension. Genome Med. 2019, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Southgate, L.; Machado, R.D.; Gräf, S.; Morrell, N.W. Molecular genetic framework underlying pulmonary arterial hypertension. Nat. Rev. Cardiol. 2020, 17, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Swietlik, E.M.; Welch, C.L.; Pauciulo, M.W.; Hagen, J.J.; Zhou, X.; Guo, Y.; Karten, J.; Pandya, D.; Tilly, T.; et al. Rare variant analysis of 4241 pulmonary arterial hypertension cases from an international consortium implicate FBLN2, PDGFD and rare de novo variants in PAH. bioRxiv 2020, 550327. [Google Scholar] [CrossRef]
- Trembath, R.C.; Thomson, J.R.; Machado, R.D.; Morgan, N.V.; Atkinson, C.; Winship, I.; Simonneau, G.; Galie, N.; Loyd, J.E.; Humbert, M.; et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N. Engl. J. Med. 2001, 345, 325–334. [Google Scholar] [CrossRef]
- Harrison, R.E.; Berger, R.; Haworth, S.G.; Tulloh, R.; Mache, C.J.; Morrell, N.W.; Aldred, M.A.; Trembath, R.C. Transforming growth factor-beta receptor mutations and pulmonary arterial hypertension in childhood. Circulation 2005, 111, 435–441. [Google Scholar] [CrossRef]
- Shintani, M.; Yagi, H.; Nakayama, T.; Saji, T.; Matsuoka, R. A new nonsense mutation of SMAD8 associated with pulmonary arterial hypertension. J. Med. Genet. 2009, 46, 331–337. [Google Scholar] [CrossRef]
- Chida, A.; Shintani, M.; Nakayama, T.; Furutani, Y.; Hayama, E.; Inai, K.; Saji, T.; Nonoyama, S.; Nakanishi, T. Missense mutations of the BMPR1B (ALK6) gene in childhood idiopathic pulmonary arterial hypertension. Circ. J. 2012, 76, 1501–1508. [Google Scholar] [CrossRef]
- Levy, M.; Eyries, M.; Szezepanski, I.; Ladouceur, M.; Nadaud, S.; Bonnet, D.; Soubrier, F. Genetic analyses in a cohort of children with pulmonary hypertension. Eur. Respir. J. 2016, 48, 1118–1126. [Google Scholar] [CrossRef]
- Wang, X.-J.; Lian, T.-Y.; Jiang, X.; Liu, S.-F.; Li, S.-Q.; Jiang, R.; Wu, W.-H.; Ye, J.; Cheng, C.-Y.; Du, Y.; et al. Germline BMP9 mutation causes idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801609. [Google Scholar] [CrossRef]
- Kerstjens-Frederikse, W.S.; Bongers, E.M.H.F.; Roofthooft, M.T.R.; Leter, E.M.; Douwes, J.M.; Van Dijk, A.; Vonk-Noordegraaf, A.; Dijk-Bos, K.K.; Hoefsloot, L.H.; Hoendermis, E.S.; et al. TBX4 mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. J. Med. Genet. 2013, 50, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Welch, C.L.; Wang, J.; Allen, P.M.; Gonzaga-Jauregui, C.; Ma, L.; King, A.K.; Krishnan, U.; Rosenzweig, E.B.; Ivy, D.D.; et al. Rare variants in SOX17 are associated with pulmonary arterial hypertension with congenital heart disease. Genome Med. 2018, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Hiraide, T.; Kataoka, M.; Suzuki, H.; Aimi, Y.; Chiba, T.; Kanekura, K.; Satoh, T.; Fukuda, K.; Gamou, S.; Kosaki, K. SOX17 mutations in Japanese patients with pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 1231–1233. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, M.S.; Ma, L.; Zhu, N.; Qi, H.; McClenaghan, C.; Gonzaga-Jauregui, C.; Dewey, F.E.; Overton, J.D.; Reid, J.G.; Shuldiner, A.R.; et al. Loss-of-function ABCC8 mutations in pulmonary arterial hypertension. Circ. Genom. Precis. Med. 2018, 11, e002087. [Google Scholar] [CrossRef]
- Eyries, M.; Montani, D.; Nadaud, S.; Girerd, B.; Levy, M.; Bourdin, A.; Trésorier, R.; Chaouat, A.; Cottin, V.; Sanfiorenzo, C.; et al. Widening the landscape of heritable pulmonary hypertension mutations in paediatric and adult cases. Eur. Respir. J. 2019, 53, 1801371. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Haarman, M.G.; Kerstjens-Frederikse, W.S.; Vissia-Kazemier, T.R.; Breeman, K.T.N.; Timens, W.; Vos, Y.J.; Roofthooft, M.T.R.; Hillege, H.L.; Berger, R.M.F. The genetic epidemiology of pediatric pulmonary arterial hypertension. J. Pediatr. 2020, 225, 65–73.e5. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Kühlbrandt, W. Biology, structure and mechanism of P-type ATPases. Nat. Rev. Mol. Cell Biol. 2004, 5, 282–295. [Google Scholar] [CrossRef]
- Madan, M.; Patel, A.; Skruber, K.; Geerts, D.; Altomare, D.A.; Iv, O.P. ATP13A3 and caveolin-1 as potential biomarkers for difluoromethylornithine-based therapies in pancreatic cancers. Am. J. Cancer. Res. 2016, 6, 1231–1252. [Google Scholar]
- Sørensen, D.M.; Holemans, T.; van Veen, S.; Martin, S.; Arslan, T.; Haagendahl, I.W.; Holen, H.W.; Hamouda, N.N.; Eggermont, J.; Palmgren, M.; et al. Parkinson disease related ATP13A2 evolved early in animal evolution. PLoS ONE 2018, 13, e0193228. [Google Scholar] [CrossRef]
- Lerche, M.; Eichstaedt, C.A.; Hinderhofer, K.; Grünig, E.; Tausche, K.; Ziemssen, T.; Halank, M.; Wirtz, H.; Seyfarth, H.-J. Mutually reinforcing effects of genetic variants and interferon-β 1a therapy for pulmonary arterial hypertension development in multiple sclerosis patients. Pulm. Circ. 2019, 9, 2045894019872192. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, J.; Swietlik, E.M.; Salmon, R.M.; Hadinnapola, C.; Nikolic, I.; Wharton, J.; Guo, J.; Liley, J.; Haimel, M.; Bleda, M.; et al. Characterization of GDF2 Mutations and Levels of BMP9 and BMP10 in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2020, 201, 575–585. [Google Scholar] [CrossRef] [PubMed]
- David, L.; Mallet, C.; Mazerbourg, S.; Feige, J.-J.; Bailly, S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 2007, 109, 1953–1961. [Google Scholar] [CrossRef] [PubMed]
- Ricard, N.; Ciais, D.; Levet, S.; Subileau, M.; Mallet, C.; Zimmers, T.A.; Lee, S.-J.; Bidart, M.; Feige, J.-J.; Bailly, S. BMP9 and BMP10 are critical for postnatal retinal vascular remodeling. Blood 2012, 119, 6162–6171. [Google Scholar] [CrossRef] [PubMed]
- Tillet, E.; Bailly, S. Emerging roles of BMP9 and BMP10 in hereditary hemorrhagic telangiectasia. Front. Genet. 2014, 5, 456. [Google Scholar] [CrossRef] [PubMed]
- Folestad, E.; Kunath, A.; Wågsäter, D. PDGF-C and PDGF-D signaling in vascular diseases and animal models. Mol. Asp. Med. 2018, 62, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.J.; Walmsley, R.; Munsey, T.S.; Smith, A.J. Receptor tyrosine kinase inhibitors cause dysfunction in adult rat cardiac fibroblasts in vitro. Toxicol. In Vitro 2019, 58, 178–186. [Google Scholar] [CrossRef]
- Morii, C.; Tanaka, H.Y.; Izushi, Y.; Nakao, N.; Yamamoto, M.; Matsubara, H.; Kano, M.R.; Ogawa, A. 3D in vitro model of vascular medial thickening in pulmonary arterial hypertension. Front. Bioeng. Biotechnol. 2020, 8, 482. [Google Scholar] [CrossRef]
- Melikyan, M.; Kareva, M.; Petraykina, E.; Averyanova, J.; Christesen, H. Hypoglycaemia in children [abstract]. In Proceedings of the 51st Annual Meeting of the European Society for Paediatric Endocrinology (ESPE), Leipzig, Germany, 20–23 September 2012. [Google Scholar]
- Timlin, M.R.; Black, A.B.; Delaney, H.M.; Matos, R.I.; Percival, C.S. Development of pulmonary hypertension during treatment with diazoxide: A case series and literature review. Pediatr. Cardiol. 2017, 38, 1247–1250. [Google Scholar] [CrossRef]
- Chen, S.C.; Dastamani, A.; Pintus, D.; Yau, D.; Aftab, S.; Bath, L.; Swinburne, C.; Hunter, L.; Giardini, A.; Christov, G.; et al. Diazoxide-induced pulmonary hypertension in hyperinsulinaemic hypoglycaemia: Recommendations from a multicentre study in the United Kingdom. Clin. Endocrinol. 2019, 91, 770–775. [Google Scholar] [CrossRef]
- Nasim, M.T.; Ogo, T.; Ahmed, M.; Randall, R.; Chowdhury, H.M.; Snape, K.M.; Bradshaw, T.Y.; Southgate, L.; Lee, G.J.; Jackson, I.; et al. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum. Mutat. 2011, 32, 1385–1389. [Google Scholar] [CrossRef] [PubMed]
- Drake, K.M.; Zygmunt, D.; Mavrakis, L.; Harbor, P.; Wang, L.; Comhair, S.A.; Erzurum, S.C.; Aldred, M.A. Altered MicroRNA processing in heritable pulmonary arterial hypertension: An important role for Smad-8. Am. J. Respir. Crit. Care Med. 2011, 184, 1400–1408. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Patient | Sex | Age of Onset | Diagnosis | Variant Identified | Mutation Category | GnomAD MAF | Variant Identifier(s) | CADD Score | SIFT Score | PolyPhen2 (HumVar) | Mutation Taster |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | M | 9 y | APAH–CHD (ASD) | ATP13A3 (exon 11) NM_024524.4 c.1148C>A (p.Thr383Lys) | Missense * | - | Novel | 32 | 0.001, D | 0.991, D | 1.0, D |
| P2 | F | 3 y | IPAH | BMP10 (exon 1) NM_014482.3 c.247G>A (p.Glu83Lys) | Missense | 0.000009 | dbSNP: rs1192957334; Not in ClinVar | 28 | 0.047, D | 0.999, D | 1.0, D |
| P3 | F | <5 y | IPAH | PDGFD (exon 4) NM_025208.5 c.550G>A (p.Glu184Lys) | Missense | - | dbSNP: rs769639743; Not in ClinVar | 28.2 | 0.007, D | 0.980, D | 0.9998, D |
| P4 | M | 19 m | IPAH | ABCC8 (exon 7) NM_000352.6 c.1069G>A (p.Val357Ile) | Missense | 0.000018 | dbSNP: rs771392416; Not in ClinVar | 23 | 0.235, T | 0.042, B | 1.0, D |
| P5 | F | 4.5 y | HPAH (mother also affected) | SMAD9 (exon 6) NM_001127217.3 c.1117G>A (p.Val373Ile) | Missense | 0.00013 | dbSNP: rs140504903; ClinVar: VCV000311894 | 27 | 0.001, D | 0.936, D | 1.0, D |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gelinas, S.M.; Benson, C.E.; Khan, M.A.; Berger, R.M.F.; Trembath, R.C.; Machado, R.D.; Southgate, L. Whole Exome Sequence Analysis Provides Novel Insights into the Genetic Framework of Childhood-Onset Pulmonary Arterial Hypertension. Genes 2020, 11, 1328. https://doi.org/10.3390/genes11111328
Gelinas SM, Benson CE, Khan MA, Berger RMF, Trembath RC, Machado RD, Southgate L. Whole Exome Sequence Analysis Provides Novel Insights into the Genetic Framework of Childhood-Onset Pulmonary Arterial Hypertension. Genes. 2020; 11(11):1328. https://doi.org/10.3390/genes11111328
Chicago/Turabian StyleGelinas, Simone M., Clare E. Benson, Mohammed A. Khan, Rolf M. F. Berger, Richard C. Trembath, Rajiv D. Machado, and Laura Southgate. 2020. "Whole Exome Sequence Analysis Provides Novel Insights into the Genetic Framework of Childhood-Onset Pulmonary Arterial Hypertension" Genes 11, no. 11: 1328. https://doi.org/10.3390/genes11111328
APA StyleGelinas, S. M., Benson, C. E., Khan, M. A., Berger, R. M. F., Trembath, R. C., Machado, R. D., & Southgate, L. (2020). Whole Exome Sequence Analysis Provides Novel Insights into the Genetic Framework of Childhood-Onset Pulmonary Arterial Hypertension. Genes, 11(11), 1328. https://doi.org/10.3390/genes11111328