Identification and Validation of Potential miRNAs, as Biomarkers for Sepsis and Associated Lung Injury: A Network-Based Approach

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sepsis-associated Acquisition of miRNA Expression Data
2.2. Differentially Expressed MicroRNA (DEM) Screening
2.3. Identification of the DEM Target Genes
2.4. Sepsis Gene Extraction from the Database
2.5. DEM–mRNA Network Construction and Hub Gene Identification
2.6. Gene Ontology and Pathway Analysis
2.7. Experimental Mice Model
2.8. miRNA Expression Validation by Real-Time Quantitative PCR
2.9. Statistical Analysis
3. Results
3.1. Sepsis-Associated DEM Identification
3.2. Prediction of Target Genes
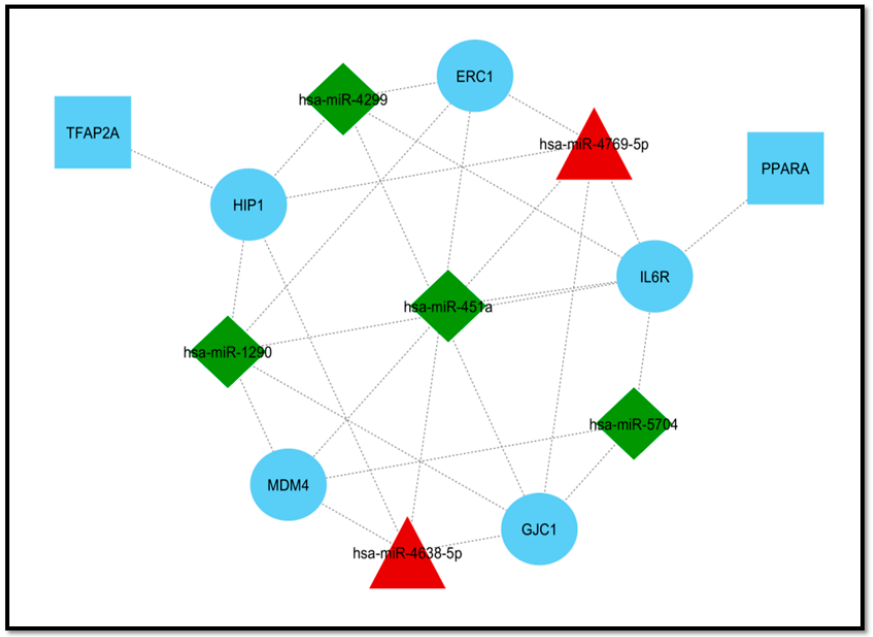
3.3. Extraction of Disease-Associated Genes and Construction of the DEM–mRNA Network
3.4. Module Detection and Pathway Enrichment Analysis
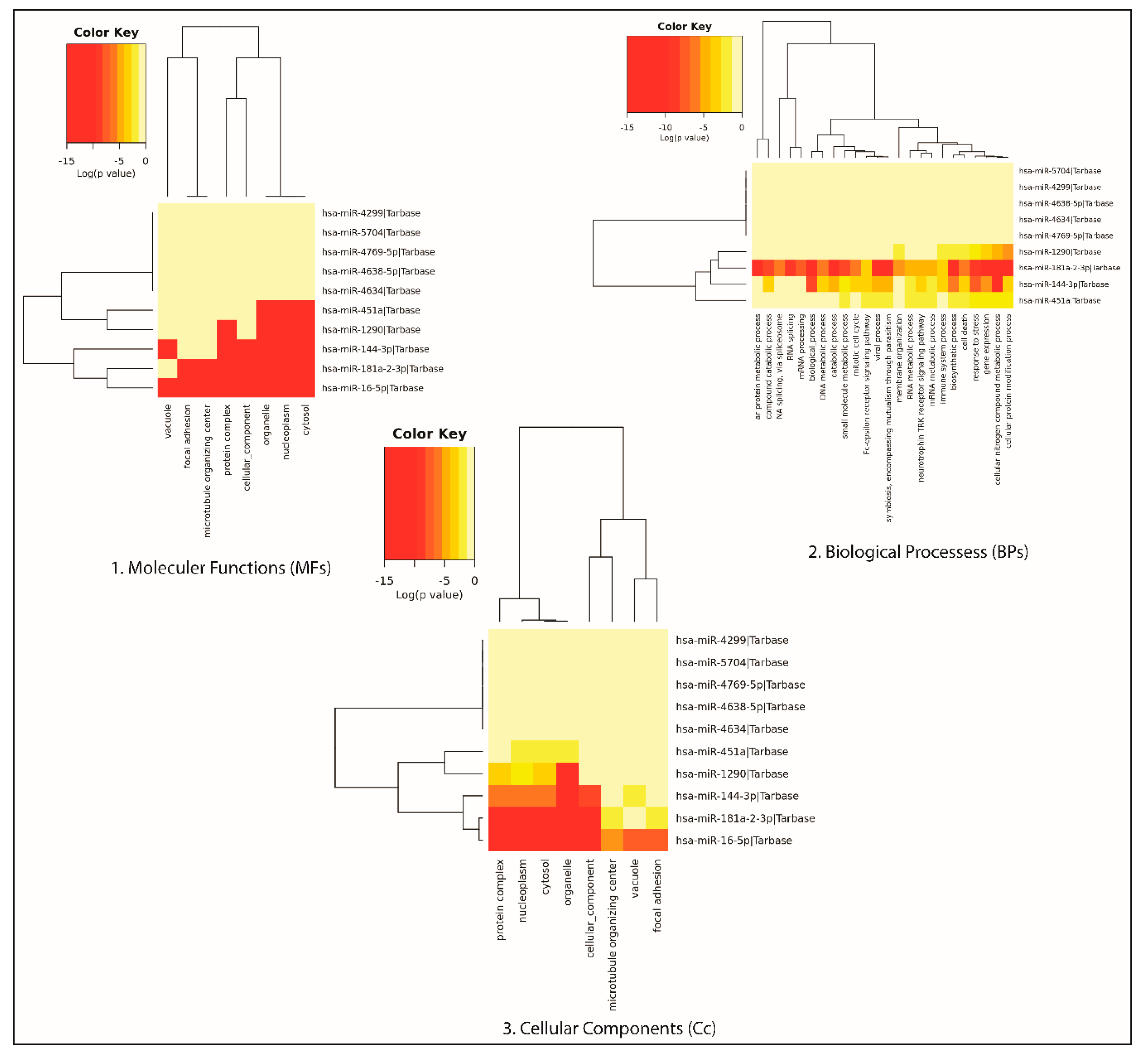
3.5. Gene Ontology of the DEMs
3.6. Validation of miRNA Expression in the Sepsis Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.C.; Shankar-Hari, M.M.; Annane, D.; Bauer, M.M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.J.; Coopersmith, C.C.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Cecconi, M.; Evans, L.; Levy, M.; Rhodes, A. Sepsis and septic shock. Lancet 2018, 392, 75–87. [Google Scholar] [CrossRef]
- Martin, G.S. Sepsis, severe sepsis and septic shock: Changes in incidence, pathogens and outcomes. Expert Rev. Anti-Infect. Ther. 2012, 10, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Walters, E. Raising Awareness for Sepsis, Sepsis Screening, Early Recognition, and Treatment in the Emergency Department. J. Emerg. Nurs. 2018, 44, 224–227. [Google Scholar] [CrossRef]
- Cohen, J. The immunopathogenesis of sepsis. Nat. Cell Biol. 2002, 420, 885–891. [Google Scholar] [CrossRef]
- Park, I.; Kim, M.; Choe, K.; Song, E.; Seo, H.; Hwang, Y.; Ahn, J.; Lee, S.-H.; Lee, J.H.; Jo, Y.H.; et al. Neutrophils disturb pulmonary microcirculation in sepsis-induced acute lung injury. Eur. Respir. J. 2019, 53, 1800786. [Google Scholar] [CrossRef]
- Angus, D.C. The Lingering Consequences of Sepsis. JAMA 2010, 304, 1833–1834. [Google Scholar] [CrossRef]
- Kumar, A.; Roberts, D.; Wood, K.E.; Light, B.; Parrillo, J.E.; Sharma, S.; Suppes, R.; Feinstein, D.; Zanotti, S.; Taiberg, L.; et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit. Care Med. 2006, 34, 1589–1596. [Google Scholar] [CrossRef]
- Hillas, G.; Vassilakopoulos, T.; Plantza, P.; Rasidakis, A.; Bakakos, P. C-reactive protein and procalcitonin as predictors of survival and septic shock in ventilator-associated pneumonia. Eur. Respir. J. 2009, 35, 805–811. [Google Scholar] [CrossRef]
- Silvestre, J.; Povoa, P.; Coelho, L.; Almeida, E.; Moreira, P.; Fernandes, A.; Mealha, R.; Sabino, H. Is C-reactive protein a good prognostic marker in septic patients? Intensive Care Med. 2009, 35, 909–913. [Google Scholar] [CrossRef]
- Fritz, H.G.; Brandes, H.; Bredle, D.L.; Bitterlich, A.; Vollandt, R.; Specht, M.; Franke, U.F.W.; Wahlers, T.; Meier-Hellmann, A. Post-operative hypoalbuminaemia and procalcitonin elevation for prediction of outcome in cardiopulmonary bypass surgery. Acta Anaesthesiol. Scand. 2003, 47, 1276–1283. [Google Scholar] [CrossRef] [PubMed]
- Wacker, C.; Prkno, A.; Brunkhorst, F.M.; Schlattmann, P. Procalcitonin as a diagnostic marker for sepsis: A systematic review and meta-analysis. Lancet Infect. Dis. 2013, 13, 426–435. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Merkel, W.; Bergmann, A. Benzodiazepin--an anti-epileptic agent with superior efficacy. MMW Munch. Med. Wochenschr 1978, 120, 120. [Google Scholar]
- Yuan, Z.; Syed, M.; Panchal, D.; Joo, M.; Bedi, C.; Lim, S.; Önyüksel, H.; Rubinstein, I.; Colonna, M.; Sadikot, R.T. TREM-1-accentuated lung injury via miR-155 is inhibited by LP17 nanomedicine. Am. J. Physiol. Cell. Mol. Physiol. 2016, 310, L426–L438. [Google Scholar] [CrossRef]
- Syed, M.; Das, P.; Pawar, A.; Aghai, Z.H.; Kaskinen, A.; Zhuang, Z.W.; Ambalavanan, N.; Pryhuber, G.; Andersson, S.; Bhandari, V. Hyperoxia causes miR-34a-mediated injury via angiopoietin-1 in neonatal lungs. Nat. Commun. 2017, 8, 1173. [Google Scholar] [CrossRef]
- Arora, S.; Dev, K.; Agarwal, B.; Das, P.; Syed, M.A. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology 2018, 223, 383–396. [Google Scholar] [CrossRef]
- Wu, X.; Yang, J.; Yu, L.; Long, D. Plasma miRNA-223 correlates with risk, inflammatory markers as well as prognosis in sepsis patients. Medicine 2018, 97, e11352. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, S.; Cao, Y.; Yang, Y. Altered miRNAs Expression Profiles and Modulation of Immune Response Genes and Proteins During Neonatal Sepsis. J. Clin. Immunol. 2014, 34, 340–348. [Google Scholar] [CrossRef]
- Szilágyi, B.; Fejes, Z.; Pócsi, M.; Kappelmayer, J.; Nagy, B., Jr. Role of sepsis modulated circulating microRNAs. EJIFCC 2019, 30, 128–145. [Google Scholar]
- Lin, Y.; Ding, Y.; Song, S.; Li, M.; Wang, T.; Guo, F. Expression patterns and prognostic value of miR-210, miR-494, and miR-205 in middle-aged and old patients with sepsis-induced acute kidney injury. Bosn. J. Basic Med. Sci. 2019, 19, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tan, L.; Tan, L.; Tian, Y.; Ma, J.; Tan, C.-C.; Wang, H.-F.; Liu, Y.; Tan, M.-S.; Jiang, T.; et al. Circulating microRNAs are promising novel biomarkers for drug-resistant epilepsy. Sci. Rep. 2015, 5, 10201. [Google Scholar] [CrossRef] [PubMed]
- Kocerha, J.; Kouri, N.; Baker, M.; Finch, N.A.; DeJesus-Hernandez, M.; Gonzalez, J.; Chidamparam, K.; Josephs, K.A.; Boeve, B.F.; Graff-Radford, N.R.; et al. Altered microRNA expression in frontotemporal lobar degeneration with TDP-43 pathology caused by progranulin mutations. BMC Genom. 2011, 12, 527. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Tan, C.; He, Y.; Zhang, G.; Xu, Y.; Tang, J. Functional miRNAs in breast cancer drug resistance. OncoTargets Ther. 2018, 11, 1529–1541. [Google Scholar] [CrossRef]
- Khalili, N.; Nouri-Vaskeh, M.; Segherlou, Z.H.; Baghbanzadeh, A.; Halimi, M.; Rezaee, H.; Baradaran, B. Diagnostic, prognostic, and therapeutic significance of miR-139-5p in cancers. Life Sci. 2020, 256, 117865. [Google Scholar] [CrossRef]
- Kumarasamy, C.; Madhav, M.R.; Sabarimurugan, S.; Krishnan, S.; Baxi, S.; Gupta, A.; Gothandam, K.M.; Jayaraj, R. Baxi Prognostic Value of miRNAs in Head and Neck Cancers: A Comprehensive Systematic and Meta-Analysis. Cells 2019, 8, 772. [Google Scholar] [CrossRef]
- Vasu, S.; Kumano, K.; Darden, C.M.; Rahman, I.; Lawrence, M.C.; Naziruddin, B. MicroRNA Signatures as Future Biomarkers for Diagnosis of Diabetes States. Cells 2019, 8, 1533. [Google Scholar] [CrossRef]
- Condrat, C.E.; Thompson, D.C.; Barbu, M.G.; Bugnar, O.L.; Boboc, A.; Cretoiu, D.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. miRNAs as Biomarkers in Disease: Latest Findings Regarding Their Role in Diagnosis and Prognosis. Cells 2020, 9, 276. [Google Scholar] [CrossRef]
- Szilágyi, B.; Fejes, Z.; Póliska, S.; Pócsi, M.; Czimmerer, Z.; Patsalos, A.; Fenyvesi, F.; Rusznyák, Á.; Nagy, G.; Kerekes, G.; et al. Reduced miR-26b Expression in Megakaryocytes and Platelets Contributes to Elevated Level of Platelet Activation Status in Sepsis. Int. J. Mol. Sci. 2020, 21, 866. [Google Scholar] [CrossRef]
- Arora, S.; Singh, P.; Dohare, R.; Jha, R.; Syed, M.A. Unravelling host-pathogen interactions: ceRNA network in SARS-CoV-2 infection (COVID-19). Gene 2020, 762, 145057. [Google Scholar] [CrossRef]
- Singh, P.; Sharma, A.; Jha, R.; Arora, S.; Ahmad, R.; Rahmani, A.H.; Almatroodi, S.A.; Dohare, R.; Syed, M.A. Transcriptomic analysis delineates potential signature genes and miRNAs associated with the pathogenesis of asthma. Sci. Rep. 2020, 10, 13354. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Singh, P.; Rahmani, A.H.; Almatroodi, S.A.; Dohare, R.; Syed, M.A. Unravelling the Role of miR-20b-5p, CCNB1, HMGA2 and E2F7 in Development and Progression of Non-Small Cell Lung Cancer (NSCLC). Biology 2020, 9, 201. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.-M.; Huang, C.-M.; Zhu, X.-Y.; Bian, F.; Pan, S.-M. Differentially expressed miRNAs in sepsis-induced acute kidney injury target oxidative stress and mitochondrial dysfunction pathways. PLoS ONE 2017, 12, e0173292. [Google Scholar] [CrossRef] [PubMed]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Sethupathy, P.; Megraw, M.; Hatzigeorgiou, A.G. A guide through present computational approaches for the identification of mammalian microRNA targets. Nat. Methods 2006, 3, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Shih, I.-H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of Mammalian MicroRNA Targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Vejnar, C.E.; Zdobnov, E.M. miRmap: Comprehensive prediction of microRNA target repression strength. Nucleic Acids Res. 2012, 40, 11673–11683. [Google Scholar] [CrossRef] [PubMed]
- Dweep, H.; Sticht, C.; Pandey, P.; Gretz, N. miRWalk–Database: Prediction of possible miRNA binding sites by “walking” the genes of three genomes. J. Biomed. Inform. 2011, 44, 839–847. [Google Scholar] [CrossRef]
- Tokar, T.; Pastrello, C.; Rossos, A.E.M.; Abovsky, M.; Hauschild, A.-C.; Tsay, M.; Lu, R.; Jurisica, I. mirDIP 4.1—Integrative database of human microRNA target predictions. Nucleic Acids Res. 2017, 46, D360–D370. [Google Scholar] [CrossRef]
- Davis, A.P.; Wiegers, T.C.; Wiegers, J.; Johnson, R.J.; Sciaky, D.; Grondin, C.J.; Mattingly, C. Chemical-Induced Phenotypes at CTD Help Inform the Predisease State and Construct Adverse Outcome Pathways. Toxicol. Sci. 2018, 165, 145–156. [Google Scholar] [CrossRef]
- Ahmad, S.; Singh, P.; Sharma, A.; Arora, S.; Shriwash, N.; Rahmani, A.H.; Almatroodi, S.A.; Manda, K.; Dohare, R.; Syed, M.A. Transcriptome Meta-Analysis Deciphers a Dysregulation in Immune Response-Associated Gene Signatures during Sepsis. Genes 2019, 10, 1005. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Panda, S.K.; Agarwal, B.; Behera, S.; Ali, S.M.; Pulse, M.E.; Solomkin, J.S.; Opal, S.M.; Bhandari, V.; Acharya, S. Novel Chitohexaose Analog Protects Young and Aged mice from CLP Induced Polymicrobial Sepsis. Sci. Rep. 2019, 9, 2904. [Google Scholar] [CrossRef] [PubMed]
- Network, A.R.D.S.; Brower, R.G.; Matthay, M.A.; Morris, A.; Schoenfeld, D.; Thompson, B.T.; Wheeler, A. Ventilation with Lower Tidal Volumes as Compared with Traditional Tidal Volumes for Acute Lung Injury and the Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2000, 342, 1301–1308. [Google Scholar] [CrossRef]
- Palevsky, P.M.; Liu, K.D.; Brophy, P.D.; Chawla, L.S.; Parikh, C.R.; Thakar, C.V.; Tolwani, A.J.; Waikar, S.S.; Weisbord, S.D. KDOQI US Commentary on the 2012 KDIGO Clinical Practice Guideline for Acute Kidney Injury. Am. J. Kidney Dis. 2013, 61, 649–672. [Google Scholar] [CrossRef] [PubMed]
- Taheri, F.; Ebrahimi, S.O.; Shareef, S.; Reiisi, S. Regulatory and immunomodulatory role of miR-34a in T cell immunity. Life Sci. 2020, 262, 118209. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yang, H.; Lehmann, C. Inhibition of GPR 55 improves dysregulated immune response in experimental sepsis. Clin. Hemorheol. Microcirc. 2019, 70, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Reithmair, M.; Buschmann, D.; Märte, M.; Kirchner, B.; Hagl, D.; Kaufmann, I.; Pfob, M.; Chouker, A.; Steinlein, O.K.; Pfaffl, M.W.; et al. Cellular and extracellular miRNAs are blood-compartment-specific diagnostic targets in sepsis. J. Cell. Mol. Med. 2017, 21, 2403–2411. [Google Scholar] [CrossRef]
- Middleton, E.A.; Rowley, J.W.; Campbell, R.A.; Grissom, C.K.; Brown, S.M.; Beesley, S.J.; Schwertz, H.; Kosaka, Y.; Manne, B.K.; Krauel, K.; et al. Sepsis alters the transcriptional and translational landscape of human and murine platelets. Blood 2019, 134, 911–923. [Google Scholar] [CrossRef]
- Arora, S.; Ahmad, S.; Irshad, R.; Goyal, Y.; Rafat, S.; Siddiqui, N.; Dev, K.; Husain, M.; Ali, S.; Mohan, A.; et al. TLRs in pulmonary diseases. Life Sci. 2019, 233, 116671. [Google Scholar] [CrossRef]
- Huang, J.; Sun, Z.; Yan, W.; Zhu, Y.; Lin, Y.; Chen, J.; Shen, B.; Wang, J. Identification of MicroRNA as Sepsis Biomarker Based on miRNAs Regulatory Network Analysis. BioMed. Res. Int. 2014, 2014, 594350. [Google Scholar] [CrossRef]
- Dumache, R.; Rogobete, A.F.; Bedreag, O.H.; Sarandan, M.; Cradigati, A.C.; Papurica, M.; Dumbuleu, C.M.; Nartita, R.; Sandesc, D. Use of miRNAs as Biomarkers in Sepsis. Anal. Cell. Pathol. 2015, 2015, 186716. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, B.; Zhang, P.; Deng, J.; Zhao, Z.; Zhang, X.; Xiao, K.; Feng, D.; Jia, Y.; Liu, Y.; et al. Identification of four novel serum protein biomarkers in sepsis patients encoded by target genes of sepsis-related miRNAs. Clin. Sci. 2014, 126, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.-G.; Zhang, S.-M.; Zhang, Y.; Zhou, Y.-Y.; Wu, H.-B.; Xu, X.-P. MicroRNAs are dynamically regulated and play an important role in LPS-induced lung injury. Can. J. Physiol. Pharmacol. 2012, 90, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Ferruelo, A.; Peñuelas, Ó.; Lorente, J.A. MicroRNAs as biomarkers of acute lung injury. Ann. Transl. Med. 2018, 6, 34. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, F.; Yu, X.; Wang, B.; Yang, Y.; Zhou, X.; Cheng, R.; Xia, S.; Zhou, X. miR-16 inhibits NLRP3 inflammasome activation by directly targeting TLR4 in acute lung injury. Biomed. Pharmacother. 2019, 112, 108664. [Google Scholar] [CrossRef]
- Hsieh, C.-H.; Rau, C.-S.; Jeng, S.-F.; Chen, Y.-C.; Lu, T.-H.; Wu, C.-J.; Wu, Y.-C.; Tzeng, S.-L.; Yang, J.C.-S. Whole blood-derived microRNA signatures in mice exposed to lipopolysaccharides. J. Biomed. Sci. 2012, 19, 69. [Google Scholar] [CrossRef]
- Möhnle, P.; Hirschberger, S.; Hinske, L.C.; Briegel, J.; Hübner, M.; Weis, S.; Dimopoulos, G.; Bauer, M.; Giamarellos-Bourboulis, E.J.; Kreth, S. MicroRNAs 143 and 150 in whole blood enable detection of T-cell immunoparalysis in sepsis. Mol. Med. 2018, 24, 54. [Google Scholar] [CrossRef]
- Goodwin, A.J.; Guo, C.; Cook, J.A.; Wolf, B.J.; Halushka, P.V.; Fan, H. Plasma levels of microRNA are altered with the development of shock in human sepsis: An observational study. Crit. Care 2015, 19, 440. [Google Scholar] [CrossRef]
- Liu, X.-F.; Wang, R.-Q.; Hu, B.; Luo, M.-C.; Zeng, Q.-M.; Zhou, H.; Huang, K.; Dong, X.-H.; Luo, Y.-B.; Luo, Z.-H.; et al. MiR-15a contributes abnormal immune response in myasthenia gravis by targeting CXCL10. Clin. Immunol. 2016, 164, 106–113. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, P.; Chen, W.; Feng, D.; Jia, Y.; Xie, L. Serum MicroRNA Signatures Identified by Solexa Sequencing Predict Sepsis Patients’ Mortality: A Prospective Observational Study. PLoS ONE 2012, 7, e38885. [Google Scholar] [CrossRef]
- Precone, V.; Stornaiuolo, G.; Amato, A.; Brancaccio, G.; Nardiello, S.; Gaeta, G.B. Different changes in mitochondrial apoptotic pathway in lymphocytes and granulocytes in cirrhotic patients with sepsis. Liver Int. 2013, 33, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-C.; Yang, J.C.-S.; Rau, C.-S.; Chen, Y.-C.; Lu, T.-H.; Lin, M.-W.; Tzeng, S.-L.; Wu, Y.-C.; Wu, C.-J.; Hsieh, C.-H. Profiling Circulating MicroRNA Expression in Experimental Sepsis Using Cecal Ligation and Puncture. PLoS ONE 2013, 8, e77936. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, M.B.; Kao, S.C.; Edelman, J.J.; Armstrong, N.J.; Vallely, M.P.; Van Zandwijk, N.; Reid, G. Haemolysis during Sample Preparation Alters microRNA Content of Plasma. PLoS ONE 2011, 6, e24145. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, R.; Lee, Y.G.; Karpurapu, M.; Syed, M.A.; Chung, S.; Deng, J.; Jeong, J.J.; Zhao, G.; Xiao, L.; Sadikot, R.T.; et al. p47phox and reactive oxygen species production modulate expression of microRNA-451 in macrophages. Free. Radic. Res. 2014, 49, 25–34. [Google Scholar] [CrossRef][Green Version]
- Rosenberger, C.M.; Podyminogin, R.L.; Navarro, G.; Zhao, G.-W.; Askovich, P.S.; Weiss, M.J.; Aderem, A. miR-451 Regulates Dendritic Cell Cytokine Responses to Influenza Infection. J. Immunol. 2012, 189, 5965–5975. [Google Scholar] [CrossRef]
- Wang, W.X.; Danaher, R.J.; Miller, C.S.; Berger, J.R.; Nubia, V.G.; Wilfred, B.S.; Neltner, J.H.; Norris, C.M.; Nelson, P.T. Expression of miR-15/107 family microRNAs in human tissues and cultured rat brain cells. Genom. Proteom. Bioinform. 2014, 12, 19–30. [Google Scholar] [CrossRef]
- Zipprich, J.T.; Bhattacharyya, S.; Mathys, H.; Filipowicz, W. Importance of the C-terminal domain of the human GW182 protein TNRC6C for translational repression. RNA 2009, 15, 781–793. [Google Scholar] [CrossRef]
- Engedal, N.; Žerovnik, E.; Rudov, A.; Galli, F.; Olivieri, F.; Procopio, A.D.; Rippo, M.R.; Monsurrò, V.; Betti, M.; Albertini, M.C. From Oxidative Stress Damage to Pathways, Networks, and Autophagy via MicroRNAs. Oxidative Med. Cell. Longev. 2018, 2018, 1–16. [Google Scholar] [CrossRef]
- Li, X.; Li, L.; Sun, Y.; Wu, J.; Wang, G.-L. Comparison of the effect of recombinant bovine wild and mutant lipopolysaccharide-binding protein in lipopolysaccharide-challenged bovine mammary epithelial cells. Cell Stress Chaperones 2016, 21, 439–452. [Google Scholar] [CrossRef]
- Jun, S.; Lee, S.; Kim, H.-C.; Ng, C.; Schneider, A.M.; Ji, H.; Ying, H.; Wang, H.; Depinho, R.A.; Park, J.-I. PAF-Mediated MAPK Signaling Hyperactivation via LAMTOR3 Induces Pancreatic Tumorigenesis. Cell Rep. 2013, 5, 314–322. [Google Scholar] [CrossRef]
- De Araujo, M.E.G.; Stasyk, T.; Taub, N.; Ebner, H.L.; Fürst, B.; Filipek, P.; Weys, S.R.; Hess, M.W.; Lindner, H.; Kremser, L.; et al. Stability of the Endosomal Scaffold Protein LAMTOR3 Depends on Heterodimer Assembly and Proteasomal Degradation. J. Biol. Chem. 2013, 288, 18228–18242. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Liang, L.; Zhang, R.; Wei, Y.; Su, L.; Tejera, P.; Guo, Y.; Wang, Z.; Lu, Q.; Baccarelli, A.A.; et al. Whole blood microRNA markers are associated with acute respiratory distress syndrome. Intensive Care Med. Exp. 2017, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Qiu, X.; Jiang, H.; Han, Y.; Wei, D.; Liu, J. Downregulation of miR-181a protects mice from LPS-induced acute lung injury by targeting Bcl-2. Biomed. Pharmacother. 2016, 84, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Xu, H. MicroRNA-181a-5p regulates inflammatory response of macrophages in sepsis. Open Med. 2019, 14, 899–908. [Google Scholar] [CrossRef] [PubMed]
- McClure, C.; Brudecki, L.; Ferguson, D.A.; Yao, Z.Q.; Moorman, J.P.; McCall, C.E.; El Gazzar, M. MicroRNA 21 (miR-21) and miR-181b Couple with NFI-A To Generate Myeloid-Derived Suppressor Cells and Promote Immunosuppression in Late Sepsis. Infect. Immun. 2014, 82, 3816–3825. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Icli, B.; Wara, A.K.; Belkin, N.; He, S.; Kobzik, L.; Hunninghake, G.M.; Vera, M.P.; Blackwell, T.S.; Baron, R.M.; et al. MicroRNA-181b regulates NF-kappaB-mediated vascular inflammation. J. Clin. Investig. 2012, 122, 1973–1990. [Google Scholar] [CrossRef]
- Dan, C.; Jinjun, B.; Zi-Chun, H.; Lin, M.; Wei, C.; Xu, Z.; Ri, Z.; Shun, C.; Wen-Zhu, S.; Qing-Cai, J.; et al. Modulation of TNF-α mRNA stability by human antigen R and miR181s in sepsis-induced immunoparalysis. EMBO Mol. Med. 2014, 7, 140–157. [Google Scholar] [CrossRef]
- Liu, G.; Liu, W.; Guo, J. Clinical significance of miR-181a in patients with neonatal sepsis and its regulatory role in the lipopolysaccharide-induced inflammatory response. Exp. Ther. Med. 2020, 19, 1977–1983. [Google Scholar] [CrossRef]
- Qin, Y.; Guo, X.; Yu, Y.; Dong, S.; Yan, Y.; Bian, X.; Zhao, C. Screening key genes and miRNAs in sepsis by RNA-sequencing. J. Chin. Med. Assoc. 2019, 83, 41–47. [Google Scholar] [CrossRef]
- Schmidt, B.; Roessler, C.; Schumann, J. Septic-Induced microRNA Expression Modulations Are Linked to Angiogenesis, Vasomotion, and Hypoxia-Induced Processes. Adv. Exp. Med. Biol. 2018, 1072, 227–231. [Google Scholar] [CrossRef]
- Wei, J.-L.; Wu, C.-J.; Chen, J.-J.; Shang, F.-T.; Guo, S.-G.; Zhang, X.-C.; Liu, H. LncRNA NEAT1 promotes the progression of sepsis-induced myocardial cell injury by sponging miR-144-3p. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 851–861. [Google Scholar]
- Wang, W.; Zhang, Y.; Zhu, B.; Duan, T.; Xu, Q.; Wang, R.; Lu, L.; Jiao, Z. Plasma microRNA expression profiles in Chinese patients with rheumatoid arthritis. Oncotarget 2015, 6, 42557–42568. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Adyshev, D.M.; Moldobaeva, N.; Mapes, B.; Elangovan, V.; Garcia, J.G.N. MicroRNA Regulation of Nonmuscle Myosin Light Chain Kinase Expression in Human Lung Endothelium. Am. J. Respir. Cell Mol. Biol. 2013, 49, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, M.; Ding, Y.; Fan, Z.; Zhang, J.; Zhang, H.; Jiang, B.; Zhu, Y. Serum MicroRNA profile in patients with colon adenomas or cancer. BMC Med. Genom. 2017, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Gil Kim, B.; Kang, S.; Han, H.H.; Lee, J.H.; Kim, J.E.; Lee, S.H.; Cho, N.H. Transcriptome-wide analysis of compression-induced microRNA expression alteration in breast cancer for mining therapeutic targets. Oncotarget 2016, 7, 27468–27478. [Google Scholar] [CrossRef]
- Krüger, O.; Plum, A.; Kim, J.S.; Winterhager, E.; Maxeiner, S.; Hallas, G.; Kirchhoff, S.; Traub, O.; Lamers, W.H.; Willecke, K. Defective vascular development in connexin 45-deficient mice. Development 2000, 127, 4179–4193. [Google Scholar] [PubMed]
- Haupt, S.; Mejía-Hernández, J.O.; Vijayakumaran, R.; Keam, S.P.; Haupt, Y. The long and the short of it: The MDM4 tail so far. J. Mol. Cell Biol. 2019, 11, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Enoh, V.T.; Lin, S.H.; Lin, C.Y.; Toliver-Kinsky, T.; Murphey, E.D.; Varma, T.K.; Sherwood, E.R. Mice depleted of alphabeta but not gammadelta T cells are resistant to mortality caused by cecal ligation and puncture. Shock 2007, 27, 507–519. [Google Scholar] [CrossRef]
- Ducut, S.J.L.; Bottero, V.; Young, D.B.; Shevchenko, A.; Mercurio, F.; Verma, I.M. Activation of transcription factor NF-kappaB requires ELKS, an IkappaB kinase regulatory subunit. Science 2004, 304, 1963–1967. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. Immunotherapeutic implications of IL-6 blockade for cytokine storm. Immunotherapy 2016, 8, 959–970. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, P.; Chen, W.; Feng, D.; Jia, Y.; Xie, L.-X. Evidence for serum miR-15a and miR-16 levels as biomarkers that distinguish sepsis from systemic inflammatory response syndrome in human subjects. Clin. Chem. Lab. Med. 2012, 50, 1423–1428. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Adj. p-Value | p-Value | logFC | miRNA_ID | Overlapped Genes Obtained by 4 Databases |
|---|---|---|---|---|---|
| 147614 | 0.000488 | 3.6 × 10−7 | −3.44103 | hsa-miR-4299 | 2090 |
| 42866 | 0.01638 | 1.52 × 10−3 | −3.70826 | hsa-miR-451a | 45 |
| 17928 | 0.006922 | 8.51 × 10−5 | −4.70341 | hsa-miR-181a-2-3p | 309 |
| 10967 | 0.016261 | 1.46 × 10−3 | −2.96498 | hsa-miR-16-5p | 594 |
| 169211 | 0.035412 | 1.28 × 10−2 | −2.45418 | hsa-miR-5704 | 930 |
| 29802 | 0.029521 | 8.59 × 10−3 | −2.90199 | hsa-miR-144-3p | 47 |
| 168568 | 0.03527 | 1.27 × 10−2 | −2.47463 | hsa-miR-1290 | 869 |
| 169214 | 0.014676 | 1.13 × 10−3 | 1.71609 | hsa-miR-4638-5p | 235 |
| 169263 | 0.004767 | 3.24 × 10−5 | 2.08135 | hsa-miR-4634 | 22 |
| 169328 | 0.014796 | 1.17 × 10−3 | 1.88279 | hsa-miR-4769-5p | 632 |
| miRNA | Disease | Expression | References |
|---|---|---|---|
| miR-16 | Sepsis (prognostic predictor) | Upregulated | [91] |
| Sepsis (distinguish sepsis/SIRS (Systemic Inflammatory response syndrome) from healthy control | Upregulated | [60] | |
| LPS-induced acute lung injury | Downregulated | [53,54] | |
| LPS-induced lung injury (in vivo model and cell line) | Downregulated | [55] | |
| LPS-induced lung injury (lung tissue and blood samples) | Upregulated | [56] | |
| Sepsis (biomarker as T-cell-mediated immunoparalysis) | Upregulated | [57] | |
| Cirrhotic patients with bacterial infection (sepsis) | Upregulated | [61] | |
| CLP-induced sepsis mice model (blood samples) | Upregulated | [62] | |
| miR-451 | CLP-induced sepsis | Upregulation | [62] |
| miR-181 | LPS-induced acute lung injury | Upregulated | [74] |
| Sepsis | Upregulated in early sepsis and downregulated in late | [75] | |
| Acute lung injury (human blood samples) | Upregulated | [72] | |
| Sepsis | Upregulated | [77] | |
| Neonatal sepsis and LPS-induced inflammation | Downregulated | [78] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, S.; Ahmed, M.M.; Hasan, P.M.Z.; Sharma, A.; Bilgrami, A.L.; Manda, K.; Ishrat, R.; Syed, M.A. Identification and Validation of Potential miRNAs, as Biomarkers for Sepsis and Associated Lung Injury: A Network-Based Approach. Genes 2020, 11, 1327. https://doi.org/10.3390/genes11111327
Ahmad S, Ahmed MM, Hasan PMZ, Sharma A, Bilgrami AL, Manda K, Ishrat R, Syed MA. Identification and Validation of Potential miRNAs, as Biomarkers for Sepsis and Associated Lung Injury: A Network-Based Approach. Genes. 2020; 11(11):1327. https://doi.org/10.3390/genes11111327
Chicago/Turabian StyleAhmad, Shaniya, Mohd Murshad Ahmed, P. M. Z. Hasan, Archana Sharma, Anwar L. Bilgrami, Kailash Manda, Romana Ishrat, and Mansoor Ali Syed. 2020. "Identification and Validation of Potential miRNAs, as Biomarkers for Sepsis and Associated Lung Injury: A Network-Based Approach" Genes 11, no. 11: 1327. https://doi.org/10.3390/genes11111327
APA StyleAhmad, S., Ahmed, M. M., Hasan, P. M. Z., Sharma, A., Bilgrami, A. L., Manda, K., Ishrat, R., & Syed, M. A. (2020). Identification and Validation of Potential miRNAs, as Biomarkers for Sepsis and Associated Lung Injury: A Network-Based Approach. Genes, 11(11), 1327. https://doi.org/10.3390/genes11111327