Multi-Trait Genomic Prediction of Yield-Related Traits in US Soft Wheat under Variable Water Regimes

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Site Description
2.2. Plant Genetic Material and Experimental Design
2.3. Traits Measurement
2.4. Phenotypic Data Analysis
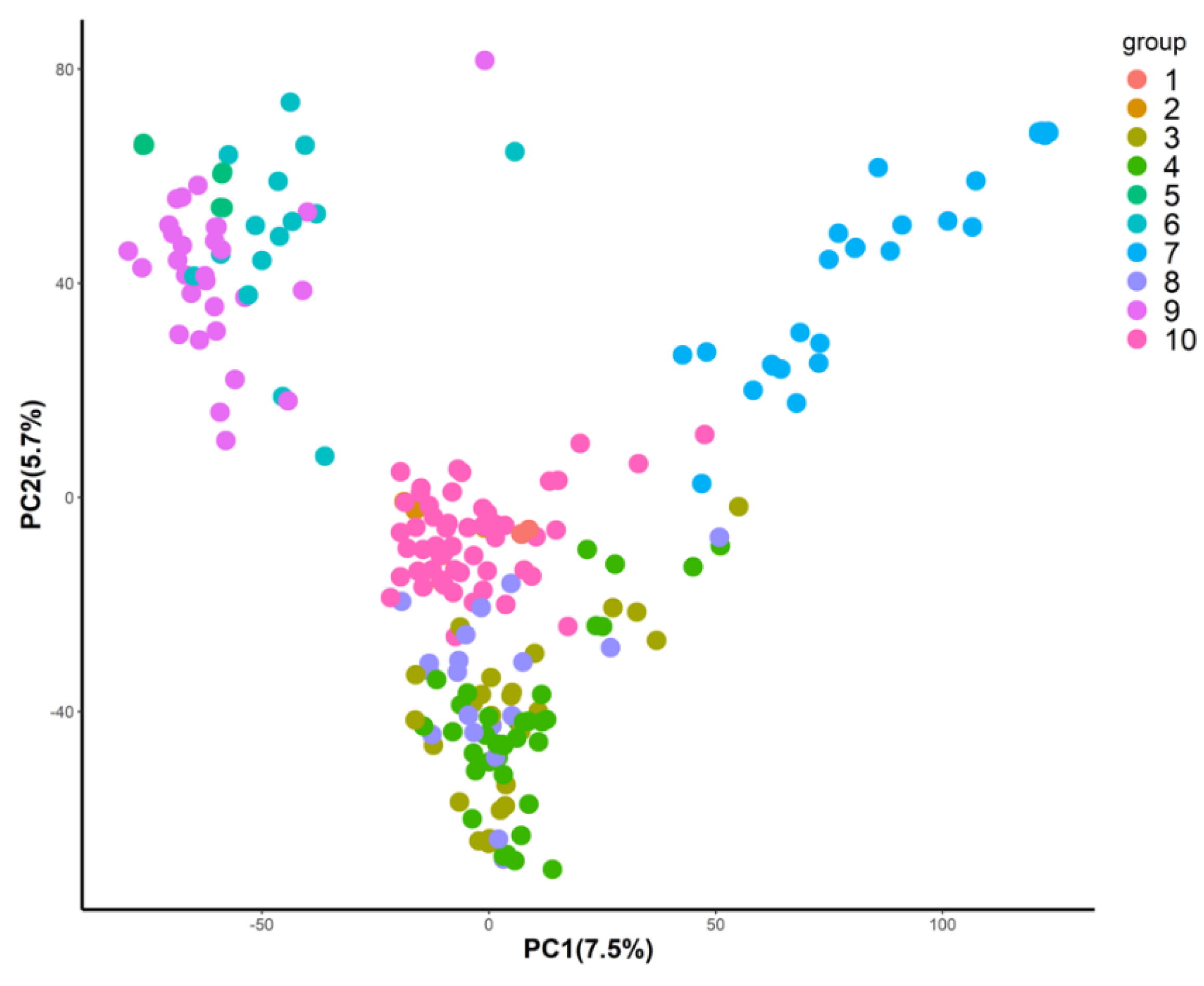
2.5. Genotypic Data Analysis
2.6. Prediction Models
2.6.1. Multi-Environment Genomic Best Linear Unbiased Predictor (MGBLUP) Model
2.6.2. Bayesian Multi-Trait Multi-Environment (BMTME) Model
2.6.3. Bayesian Multi-Output Regressor Stacking (BMORS) Model
2.6.4. Deep Learning (DL) Models
2.7. Model Evaluation
2.8. Software Implementation
2.9. Data Availability
3. Results
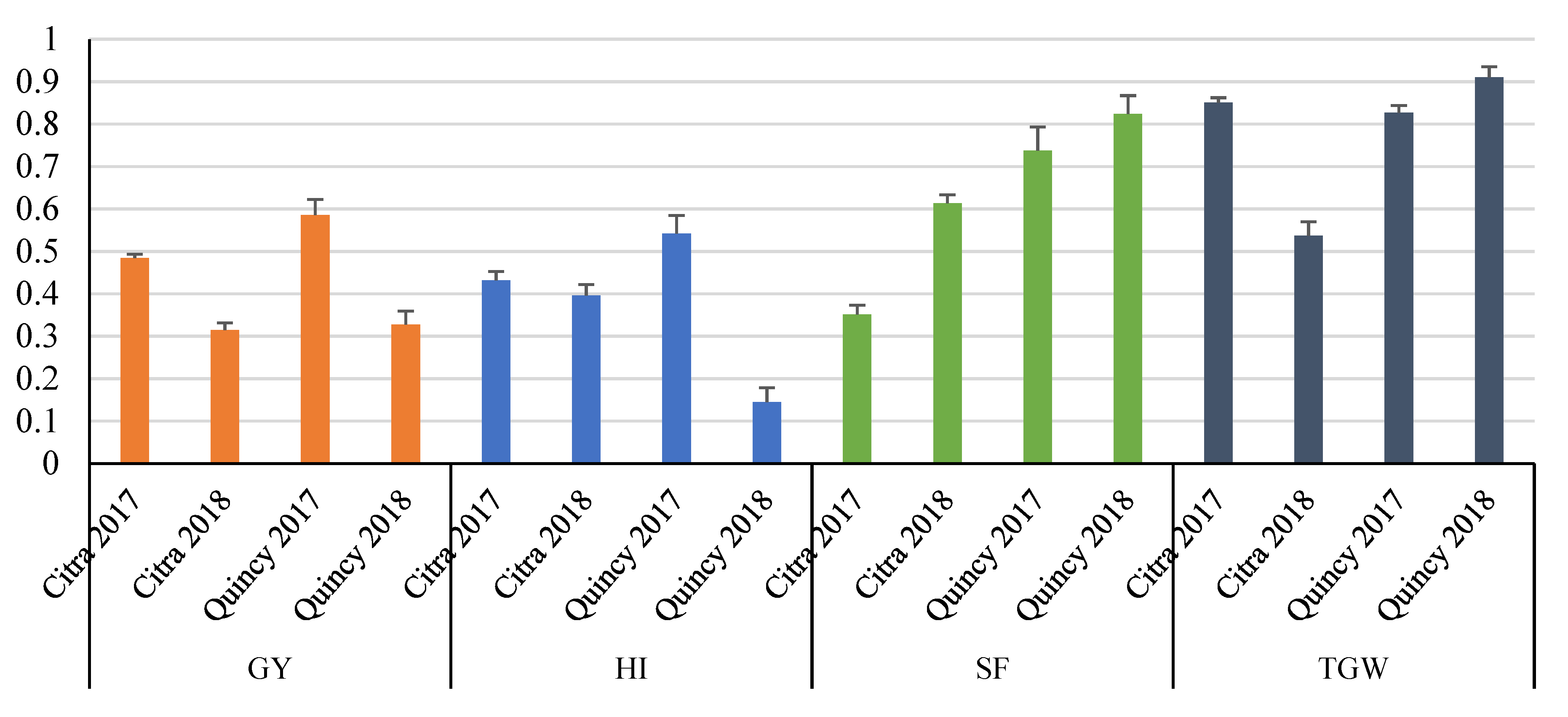
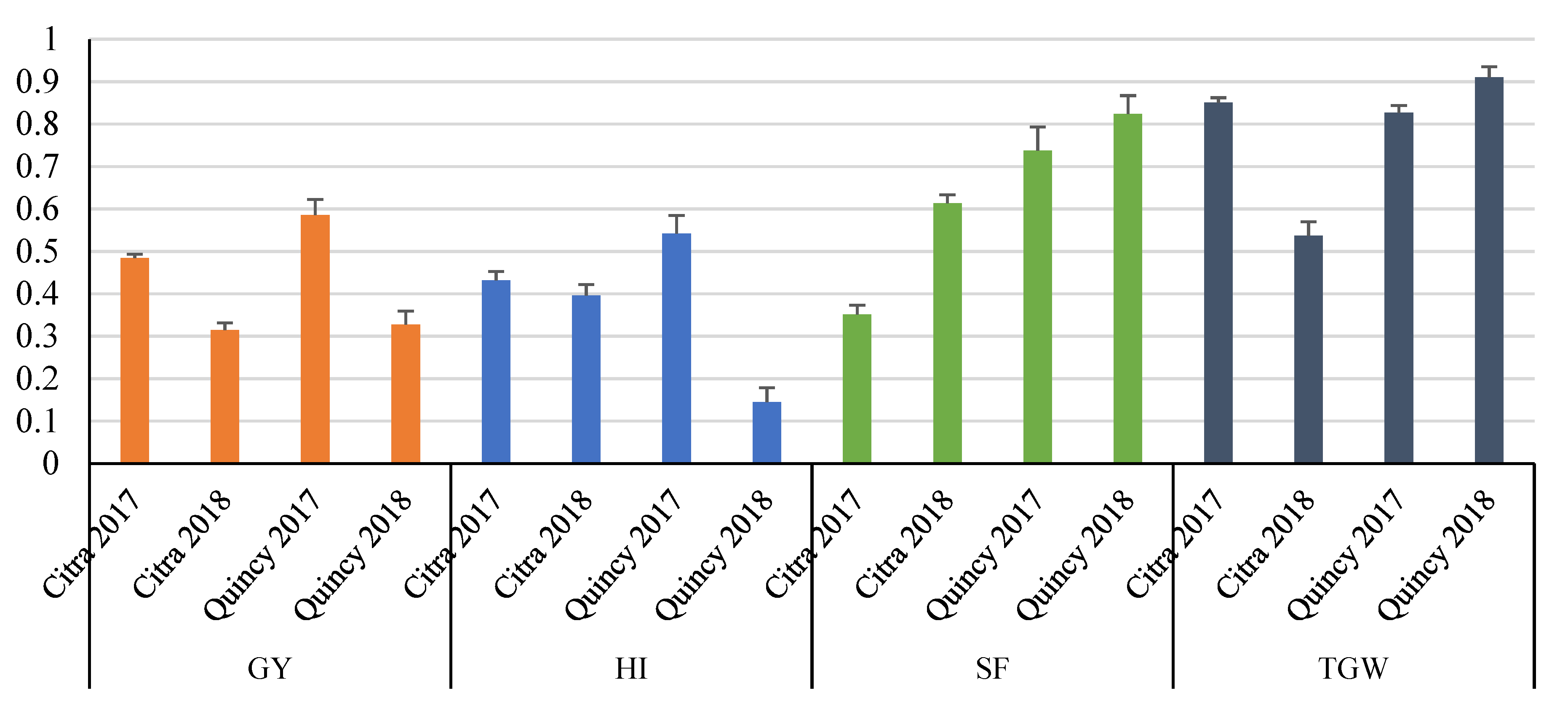
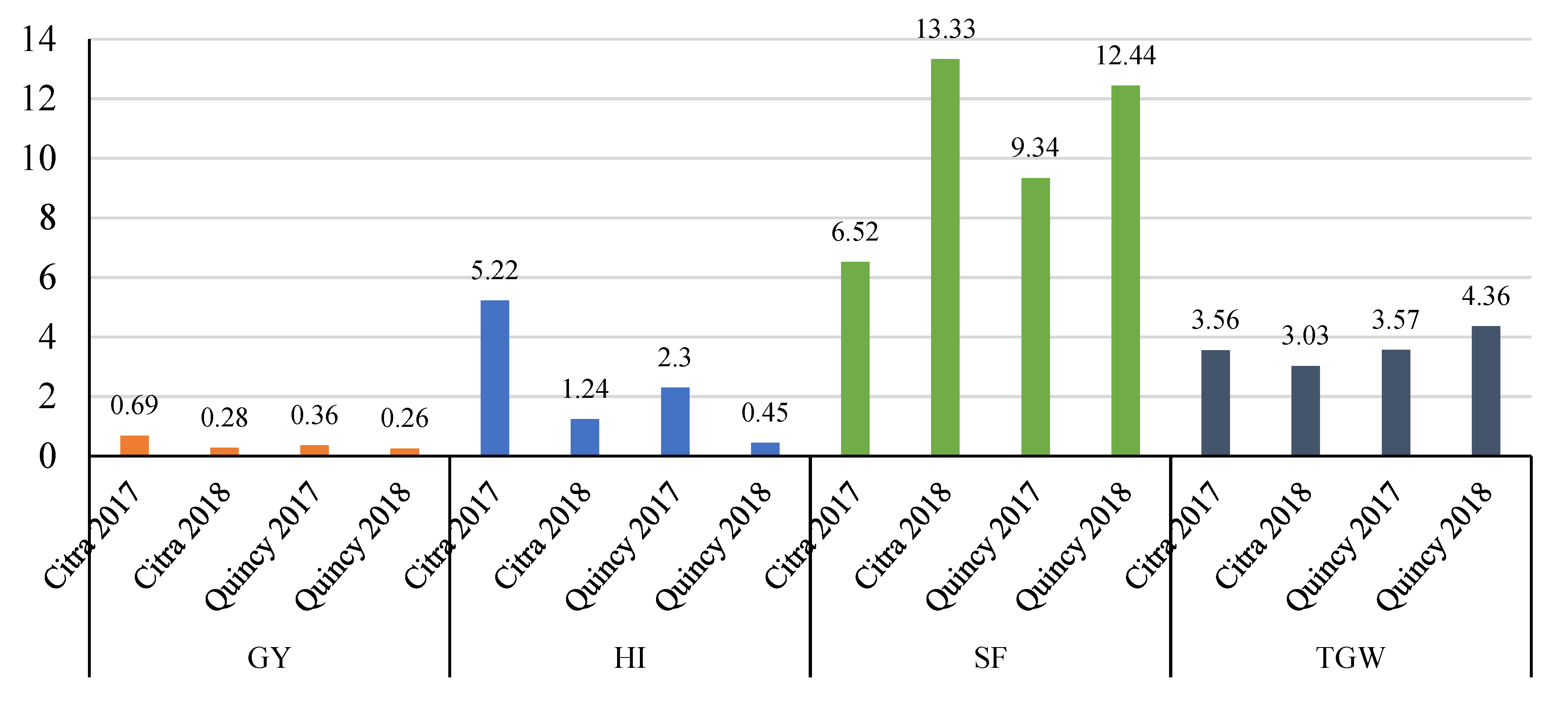
3.1. Descriptive Statistics
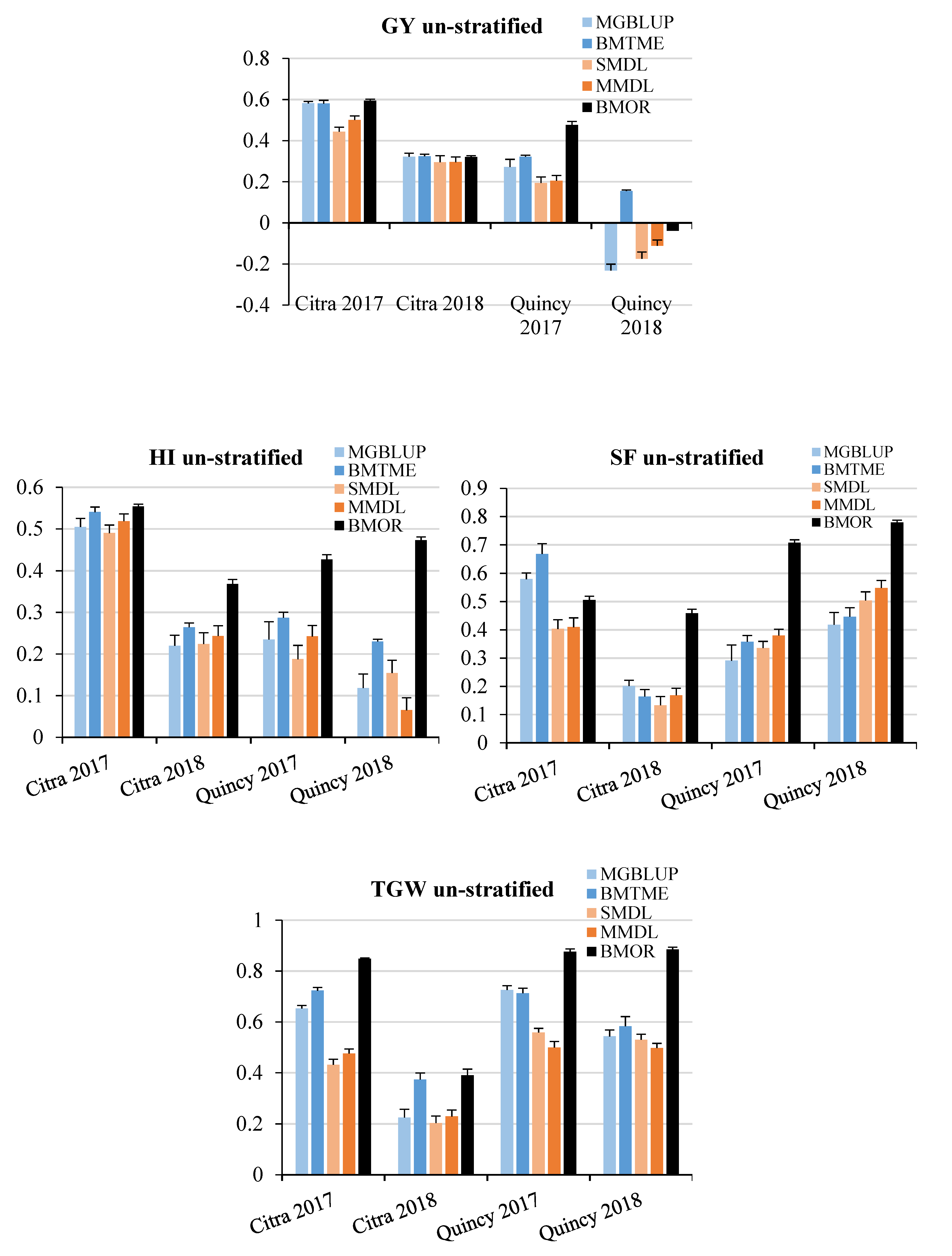
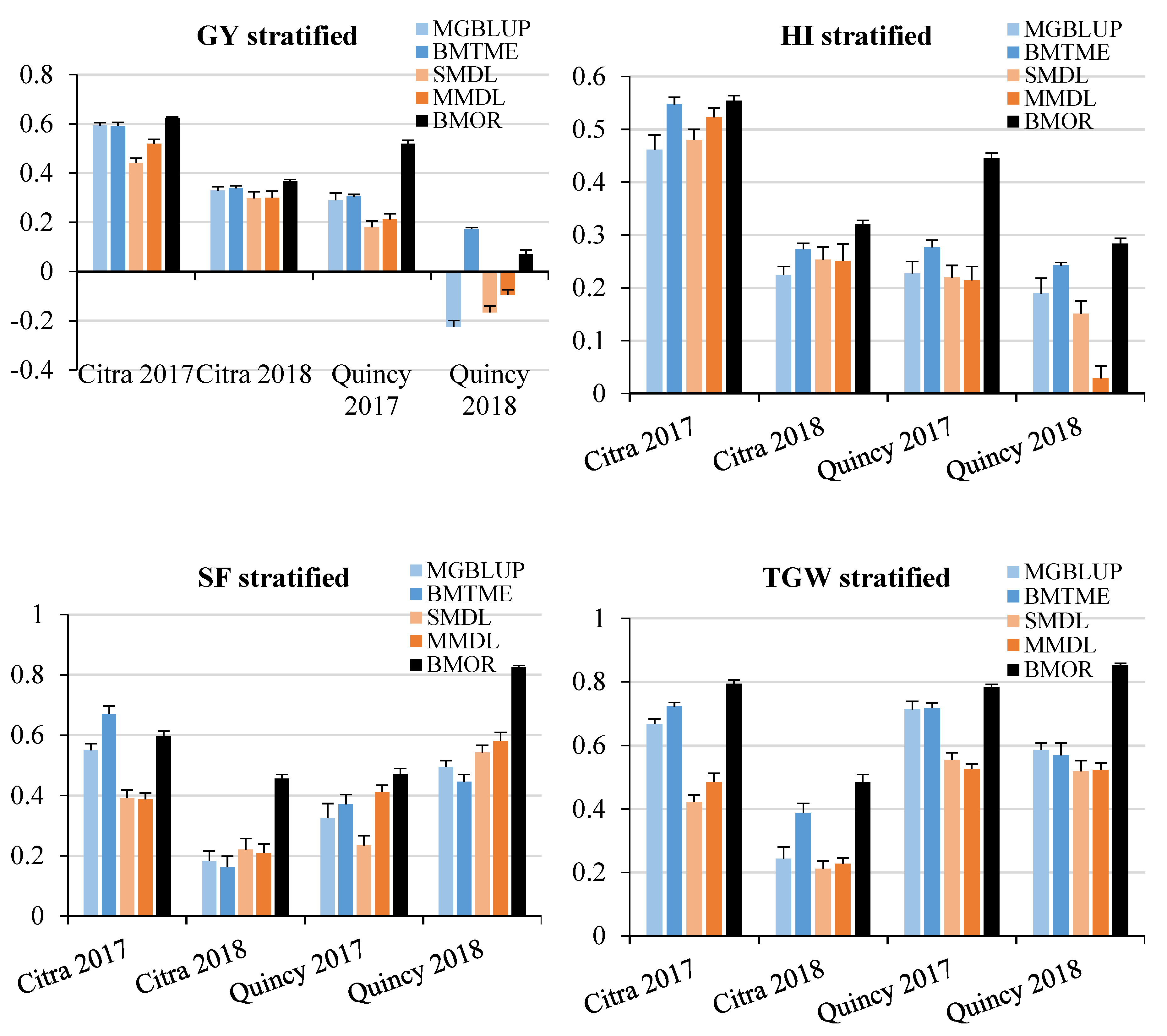
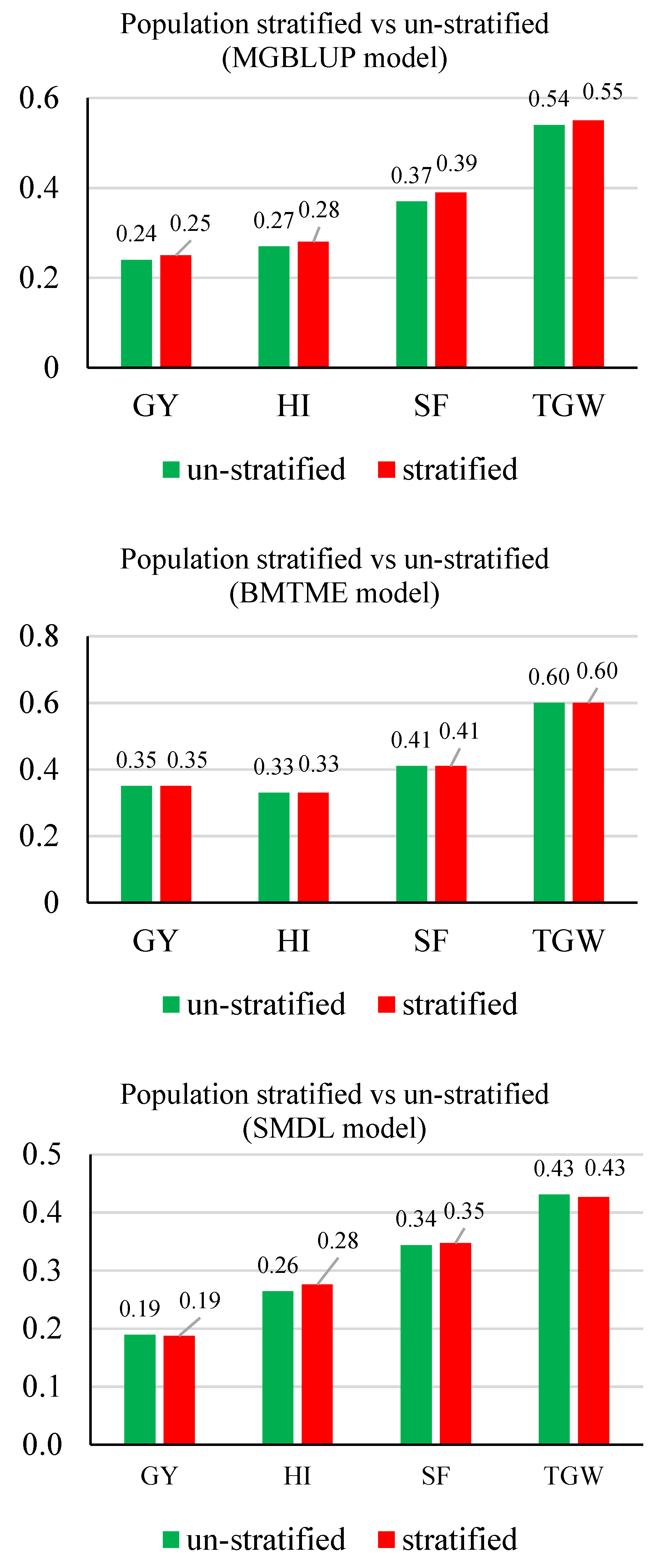
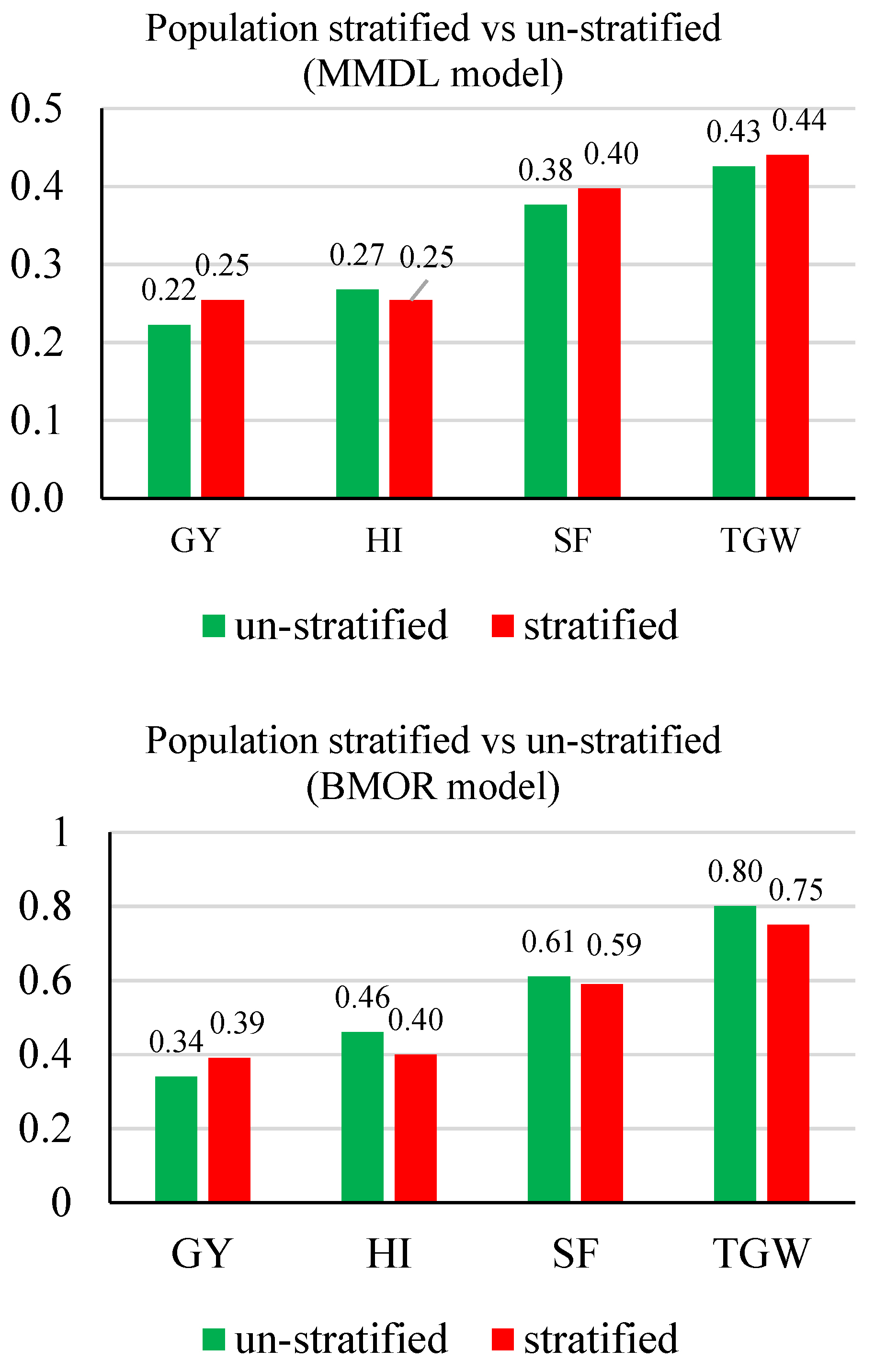
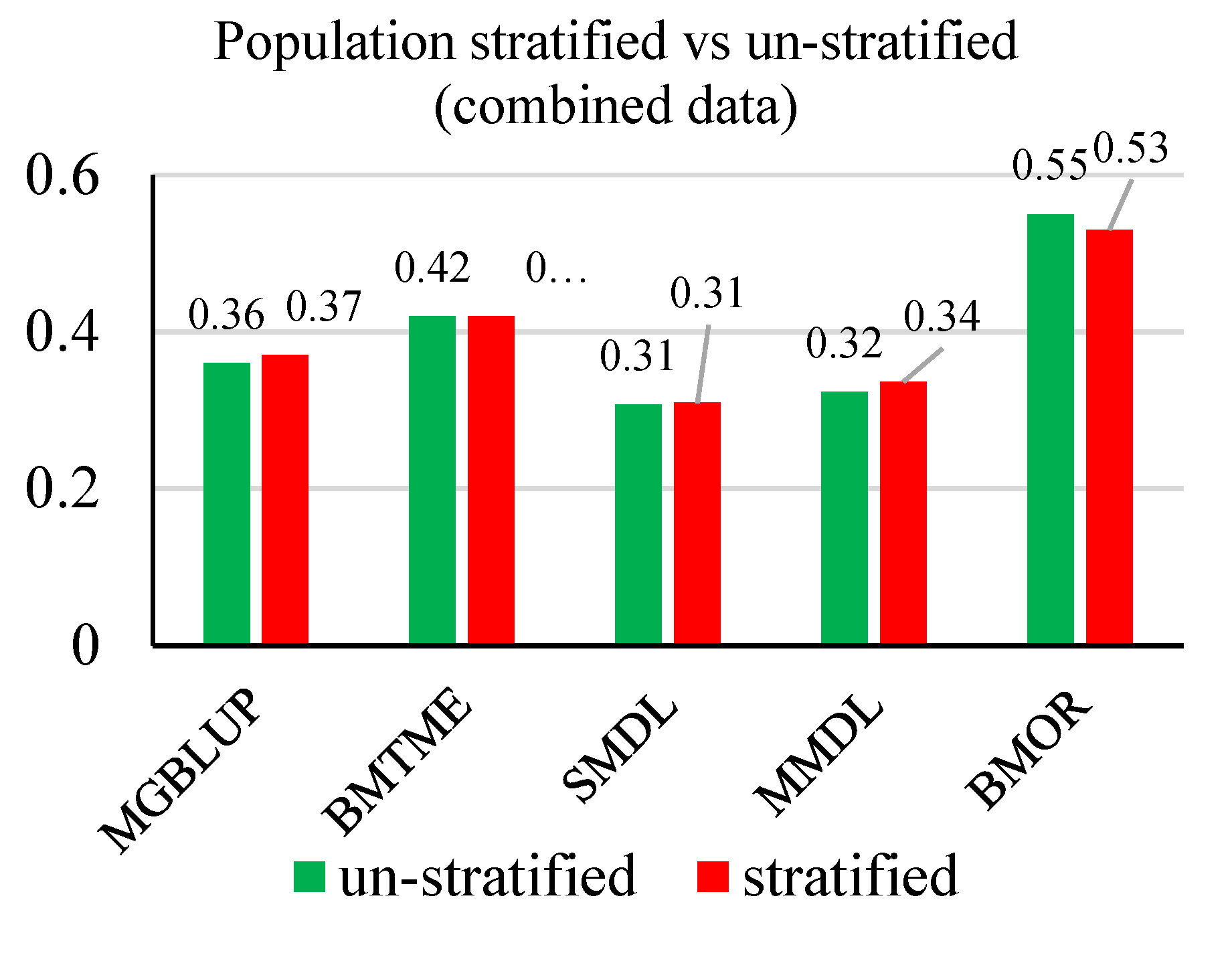
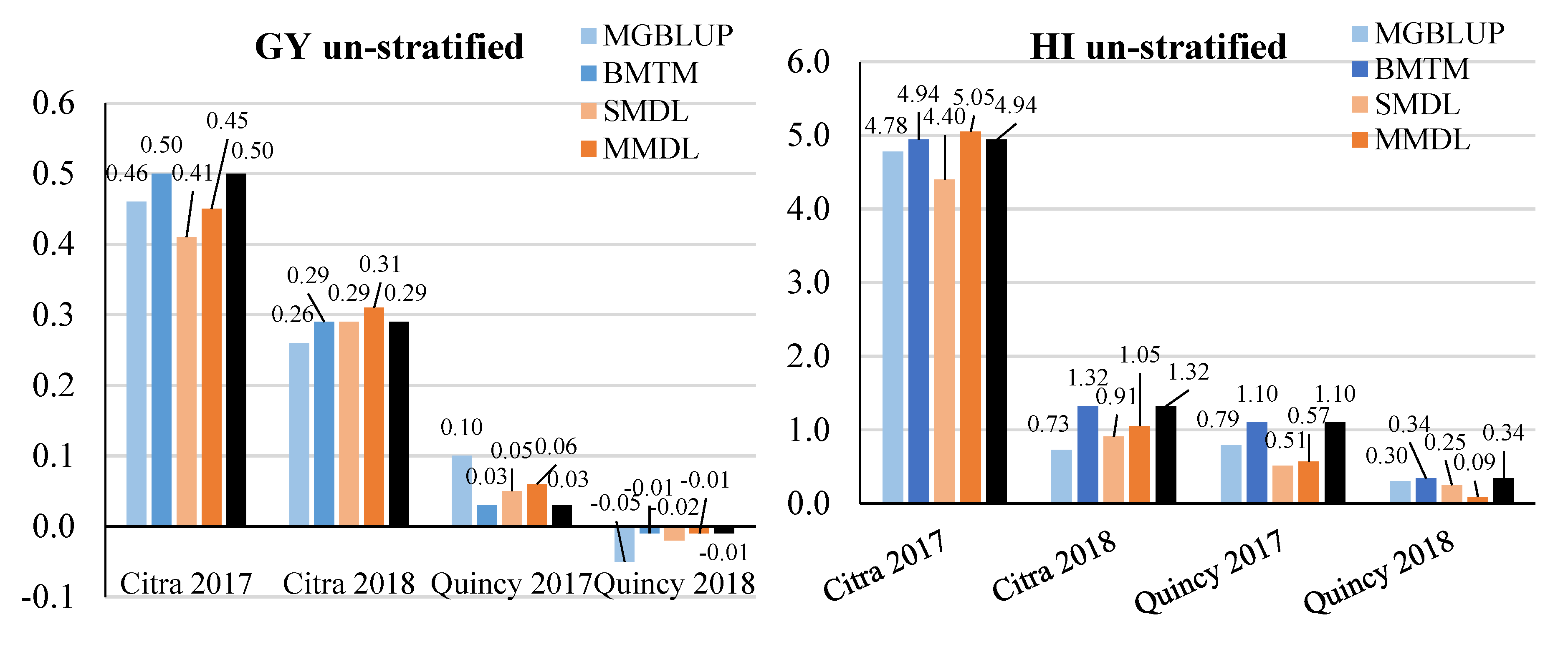
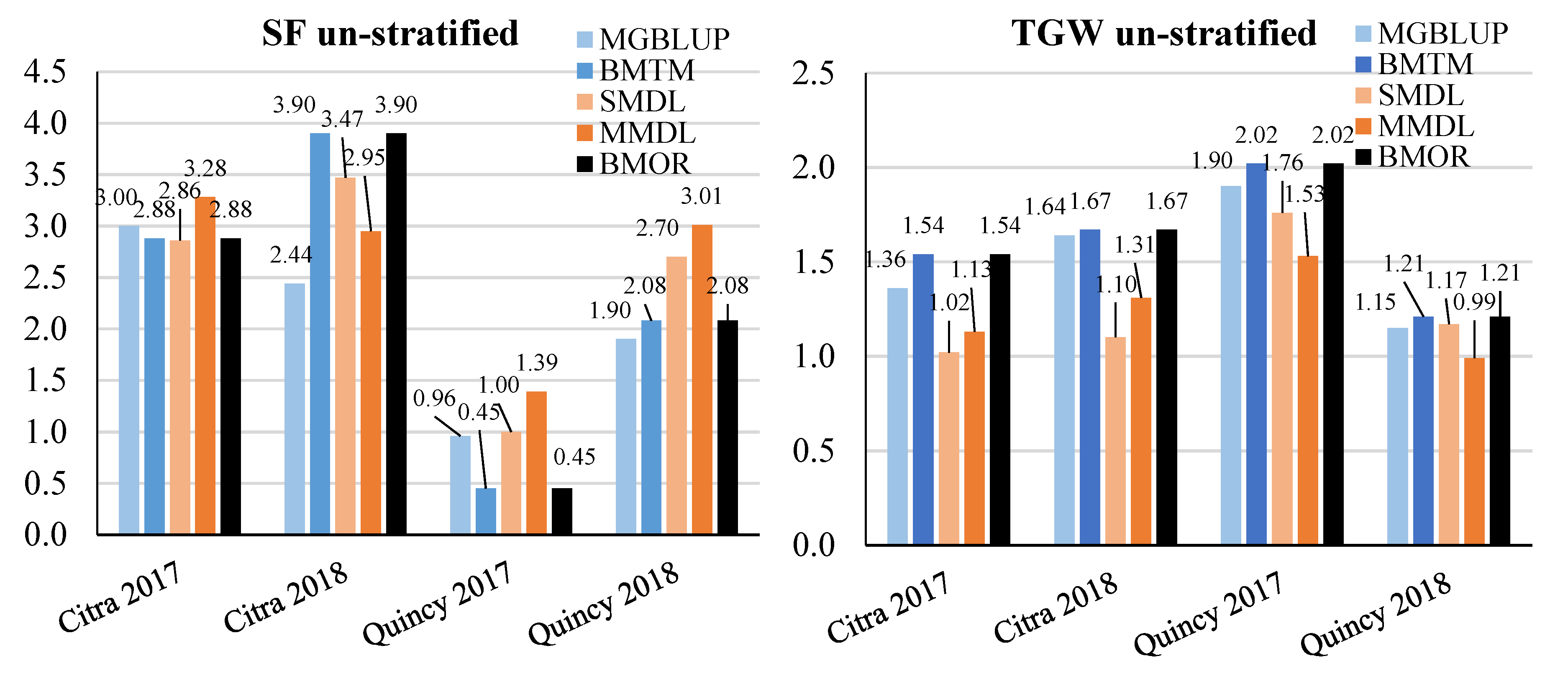
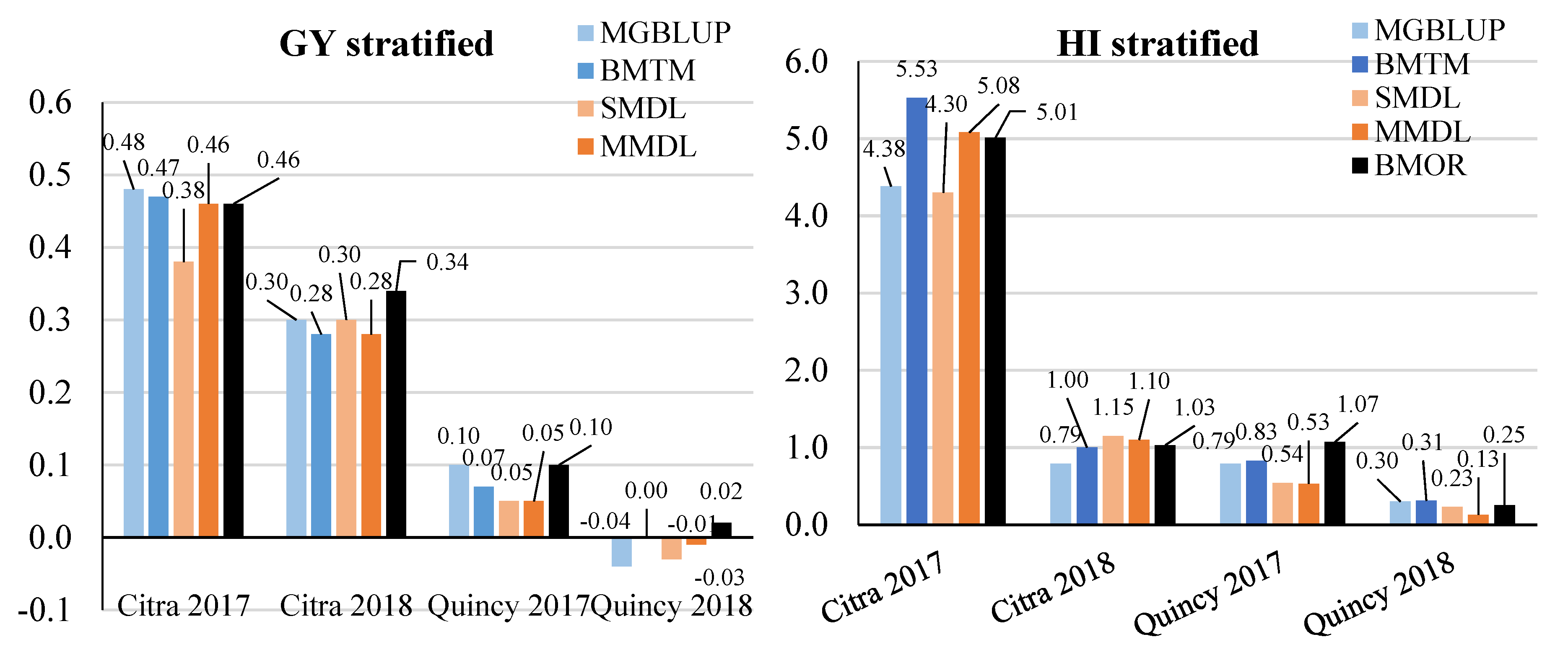
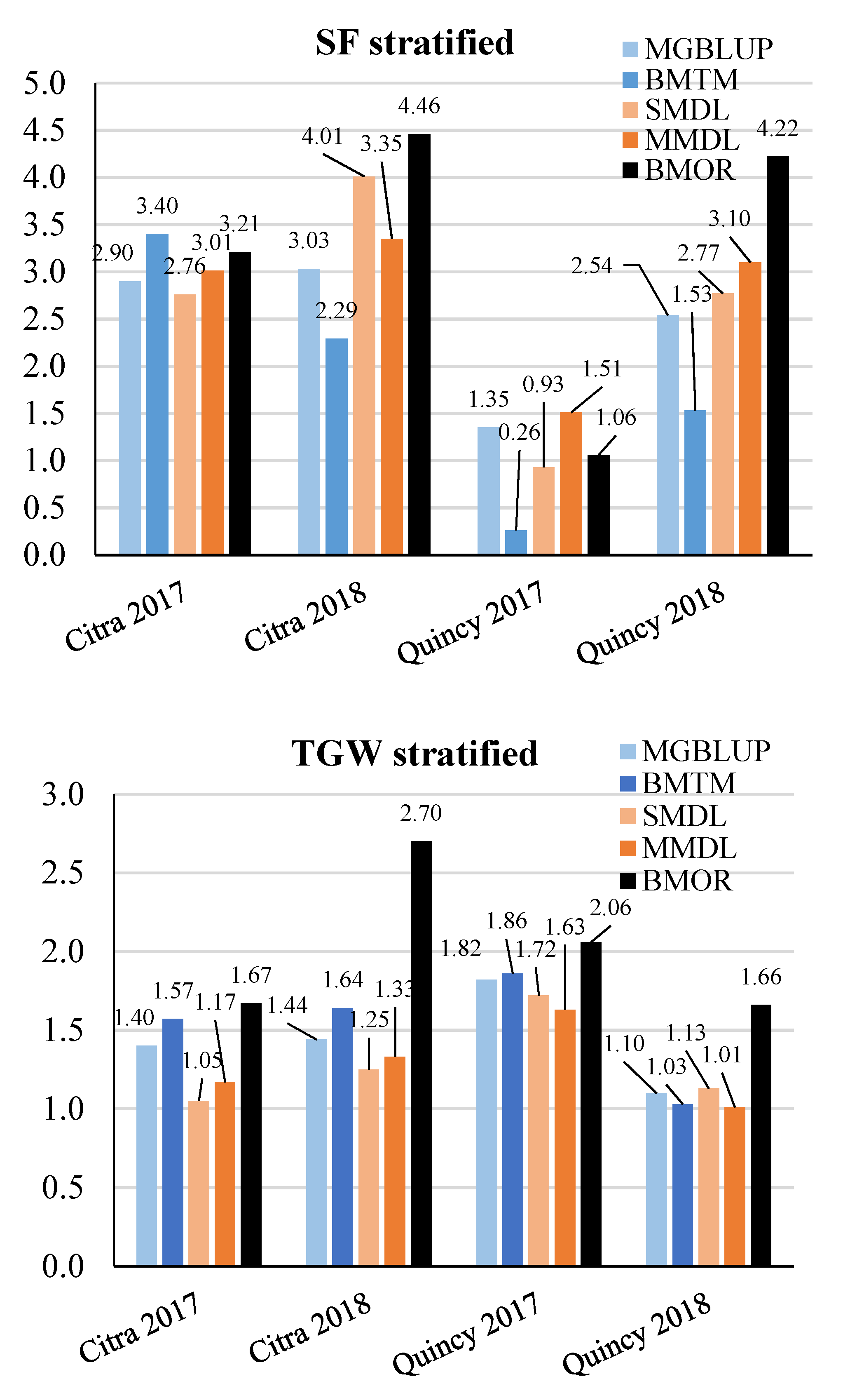
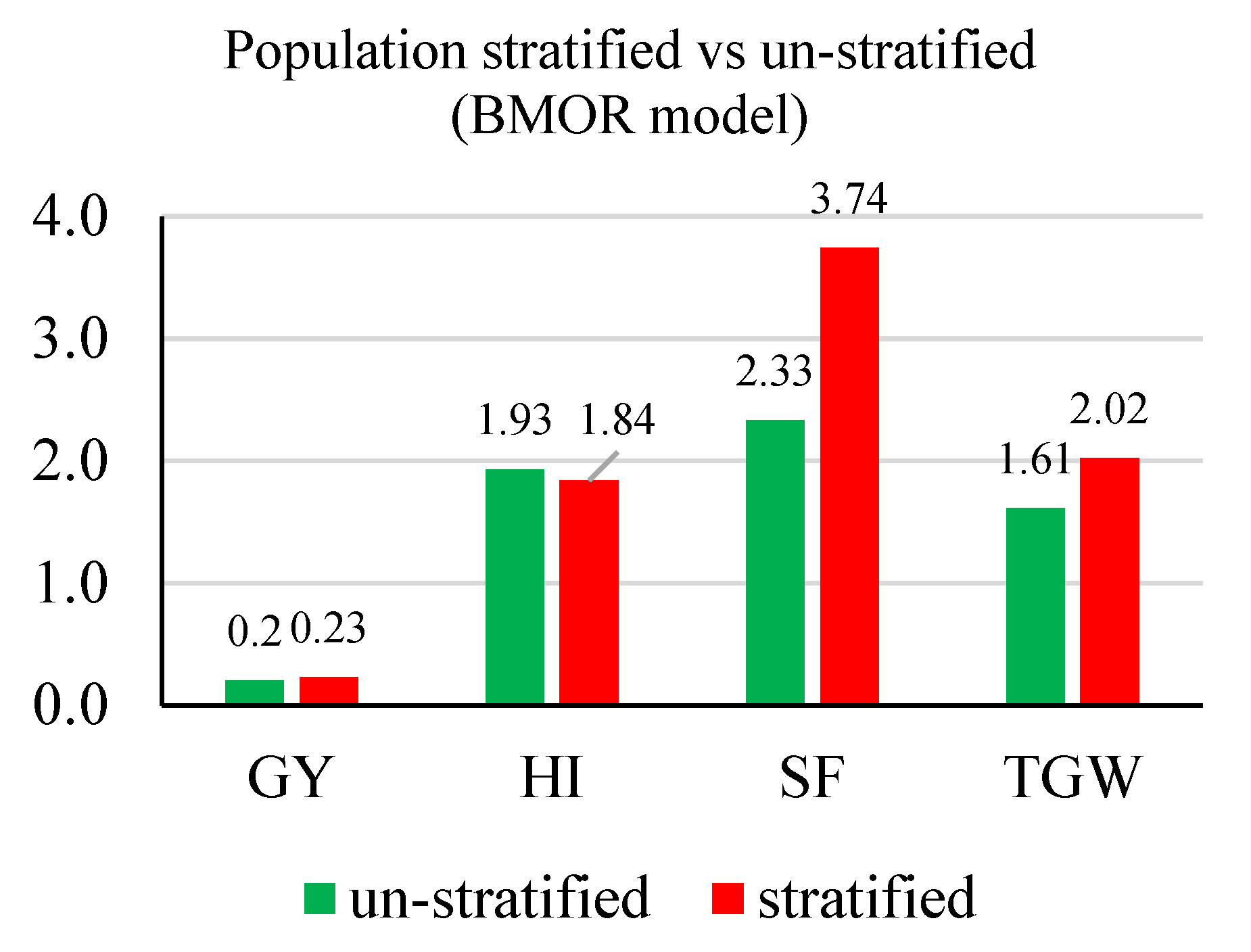
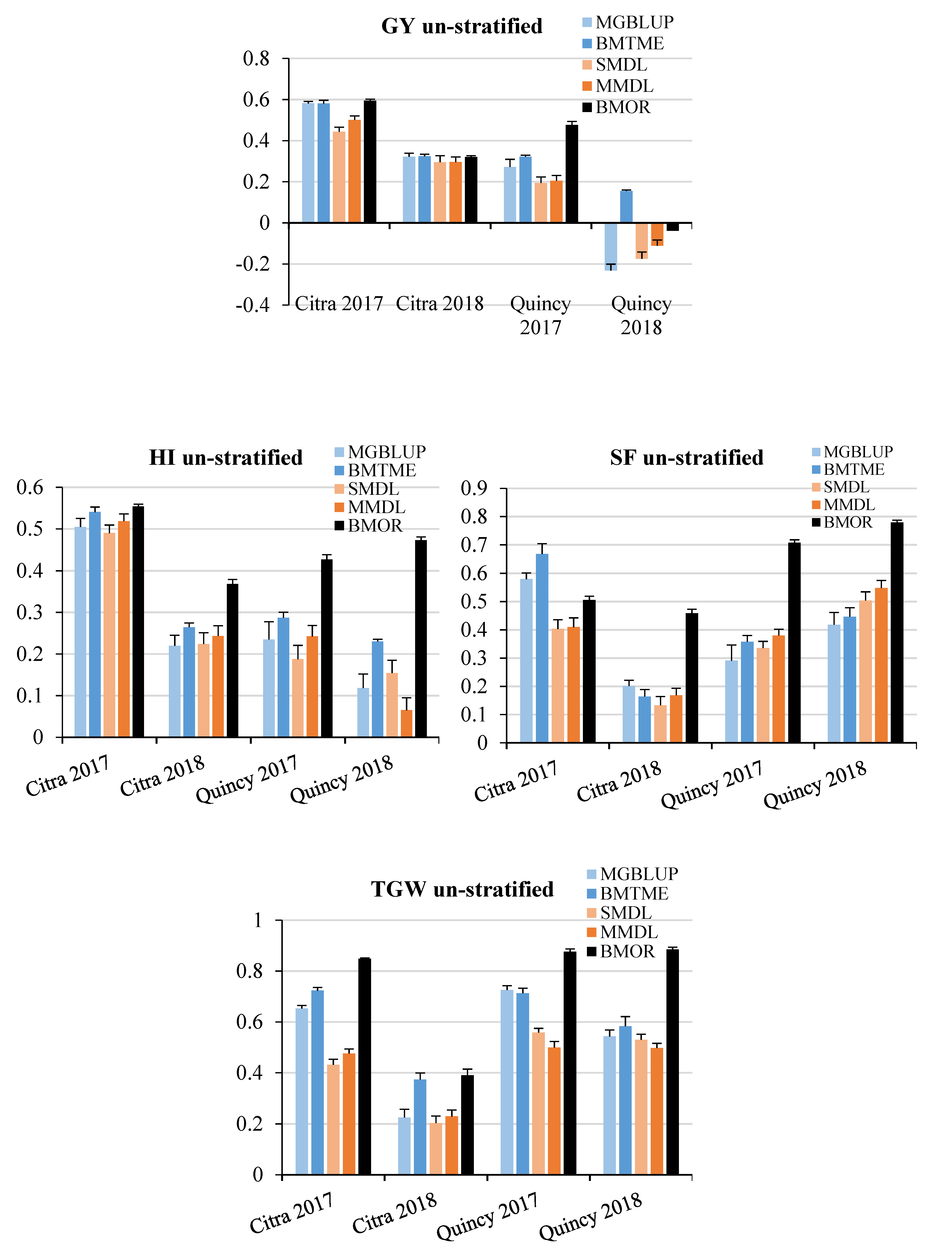
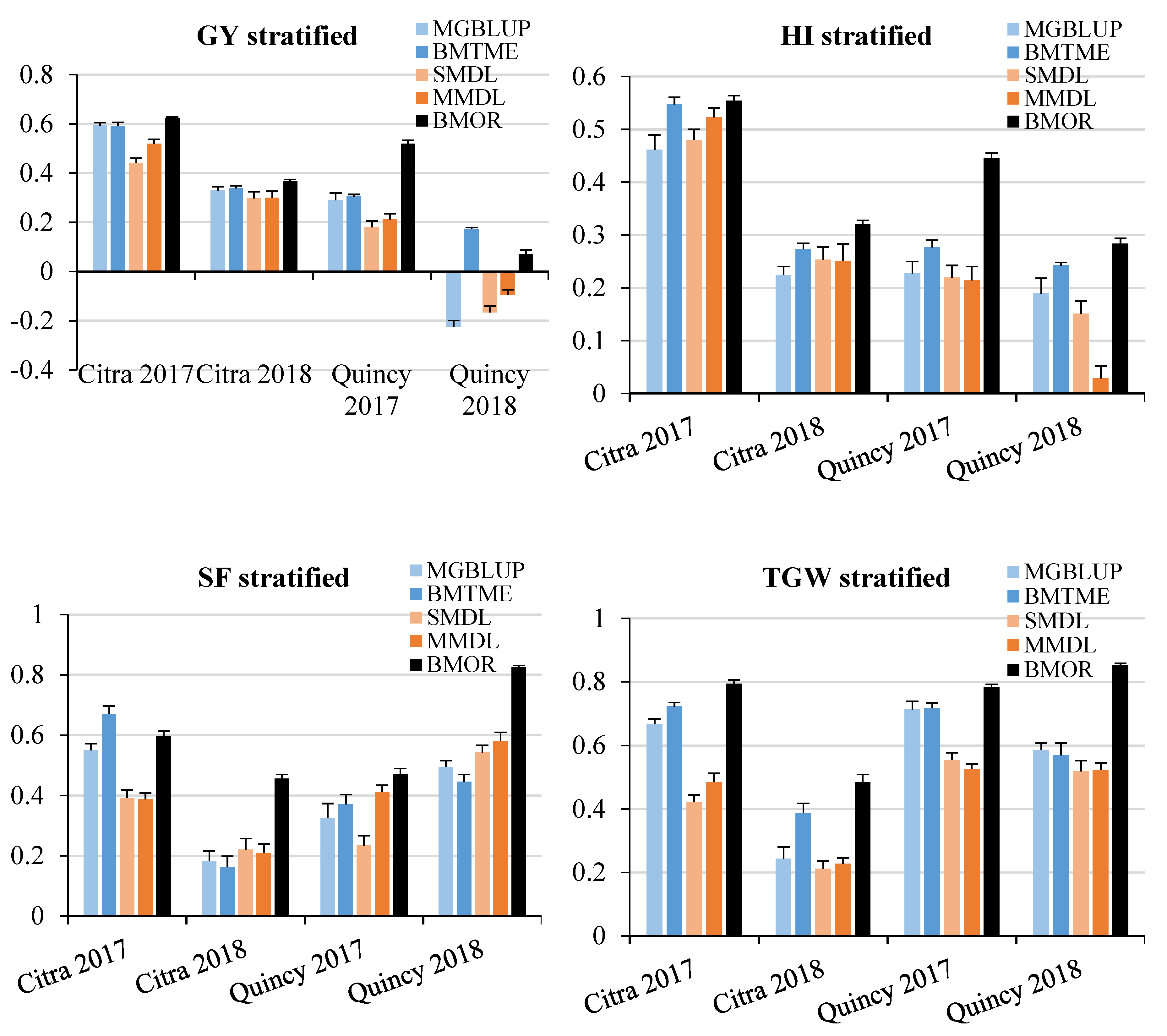
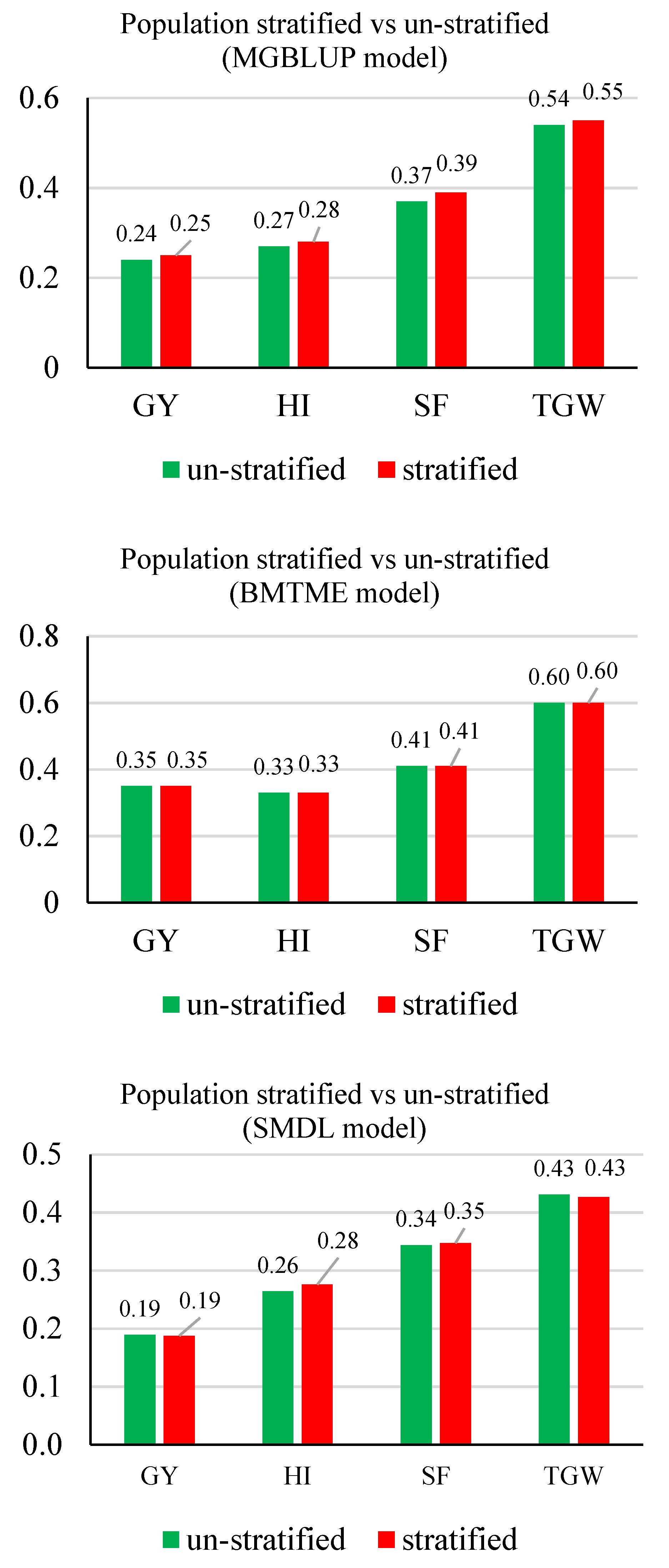
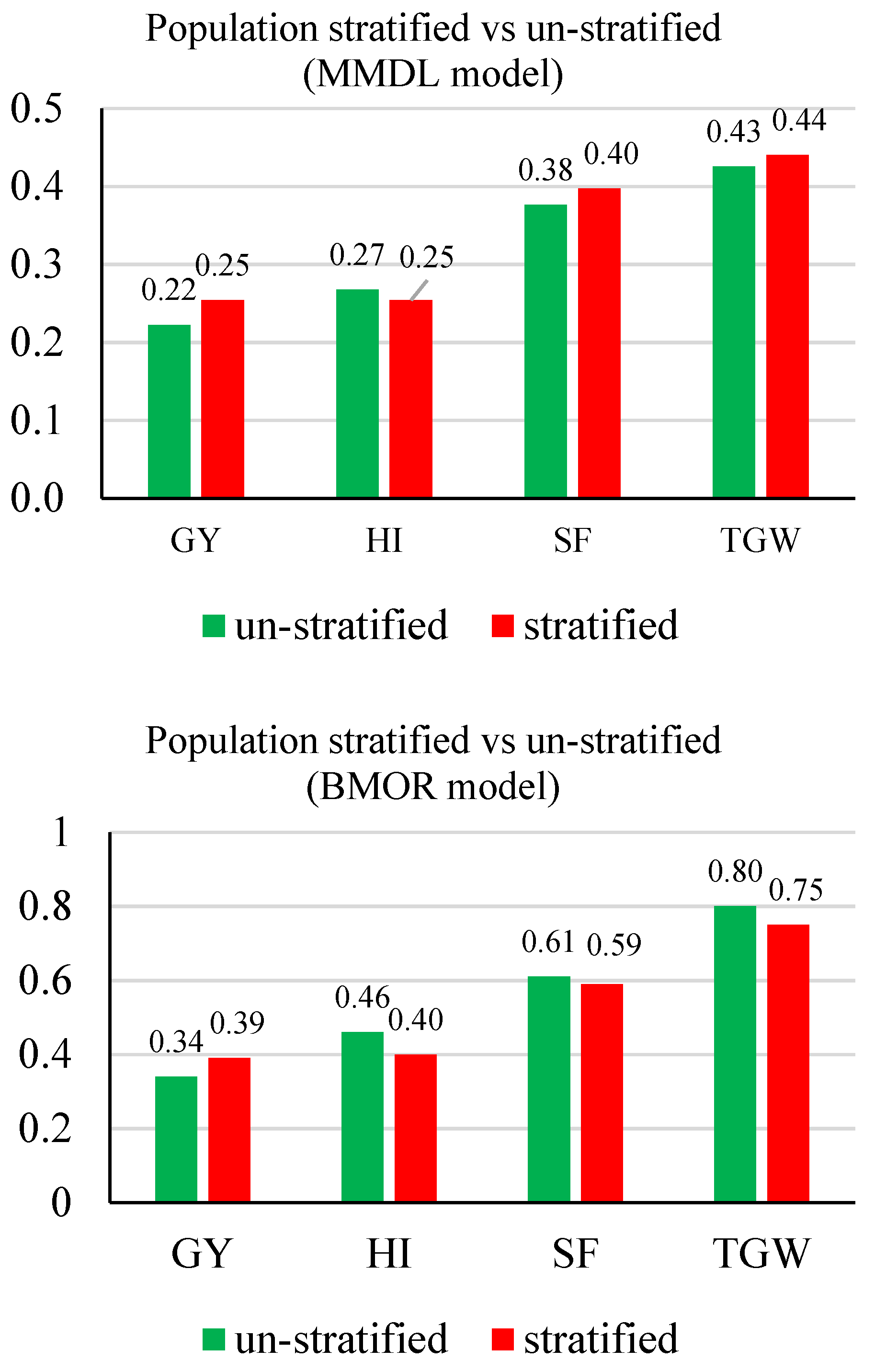
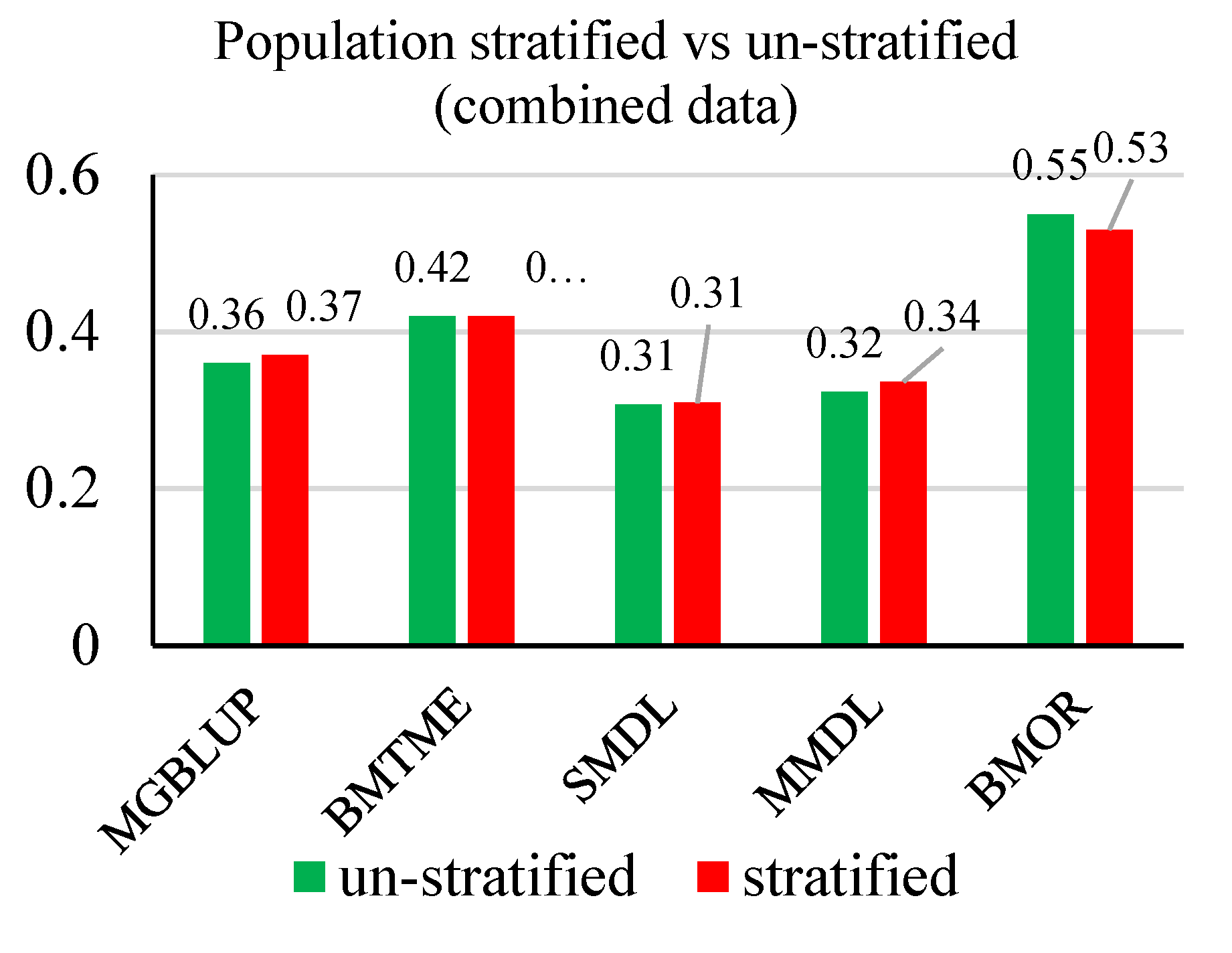
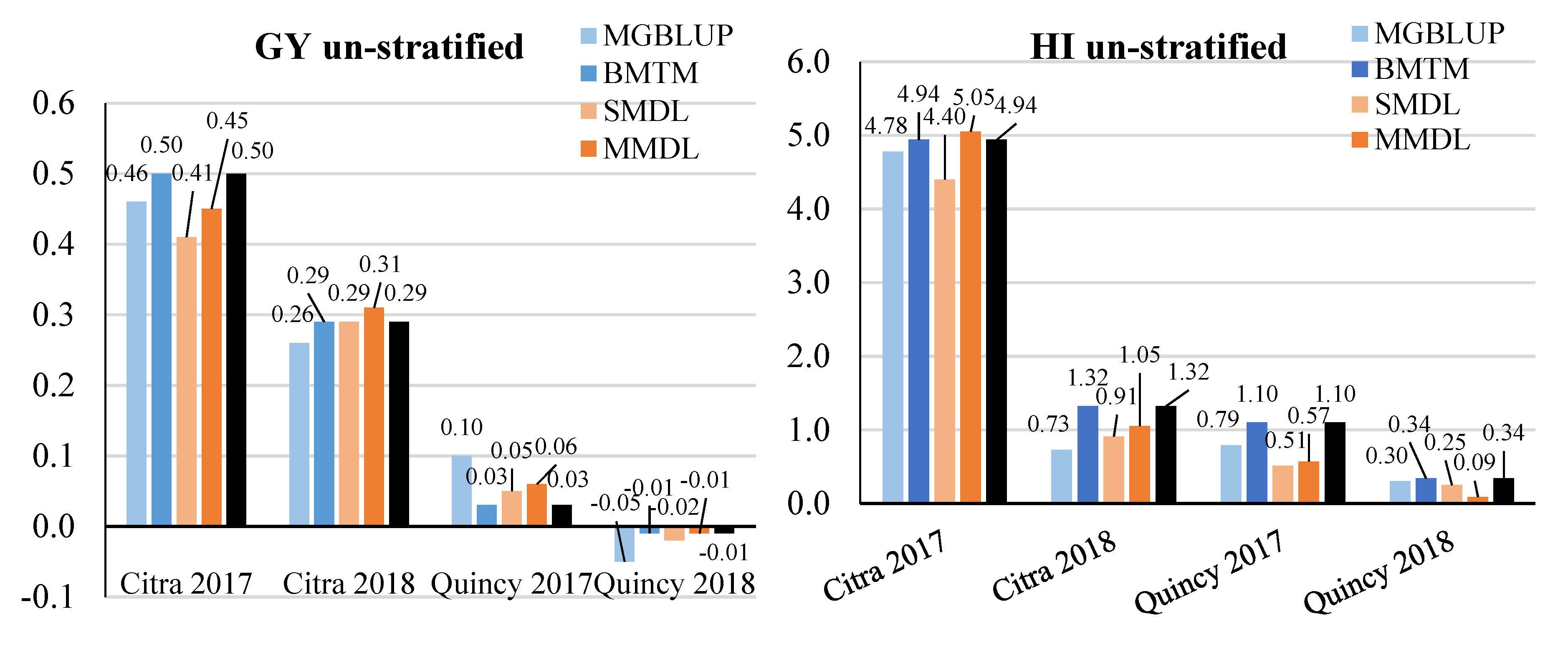
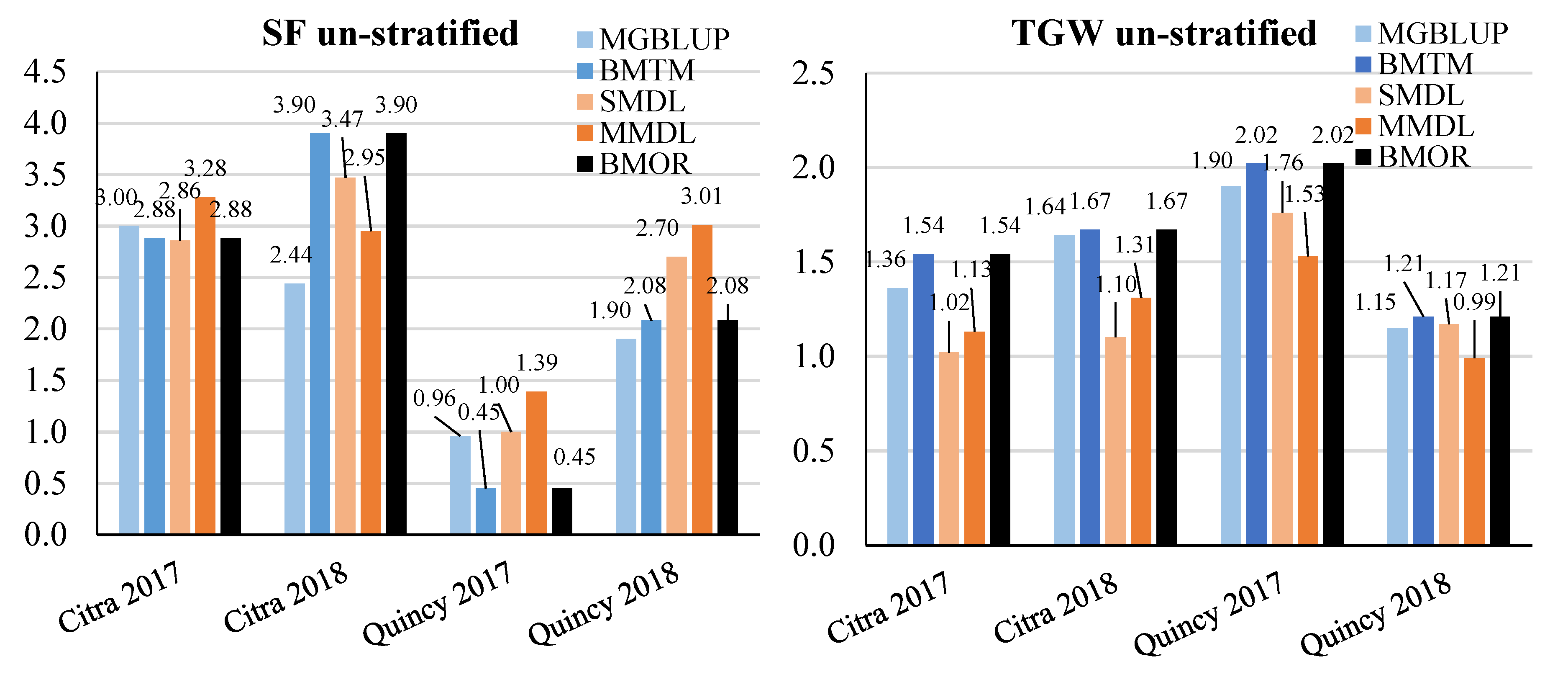
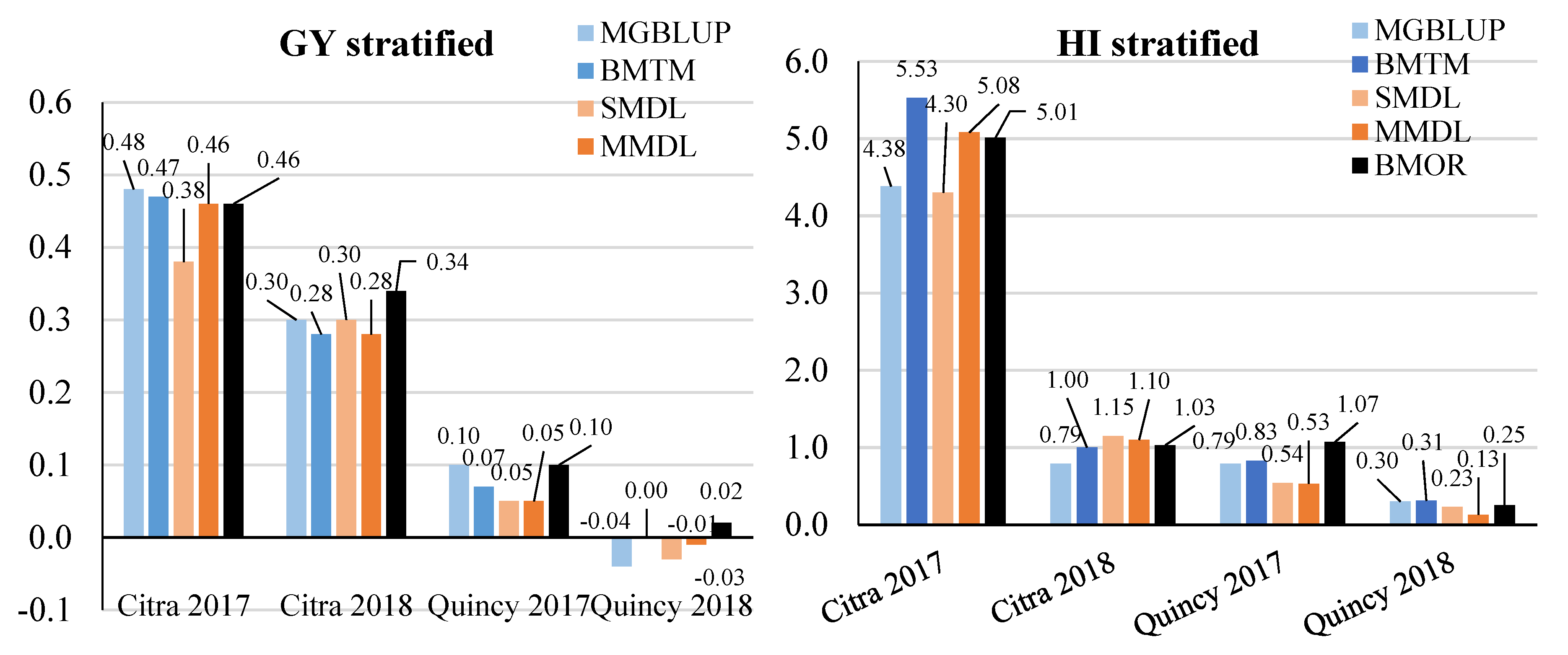
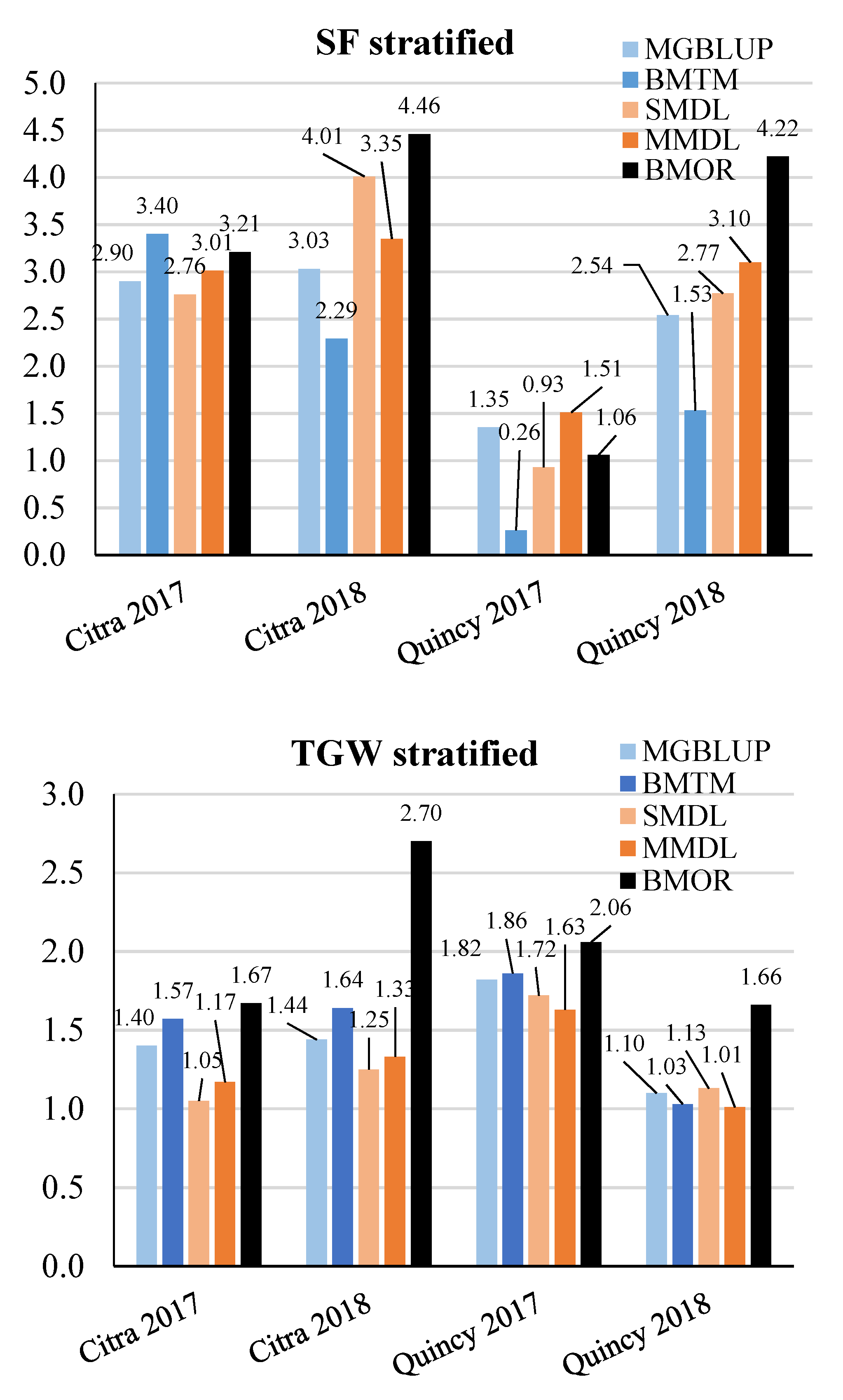
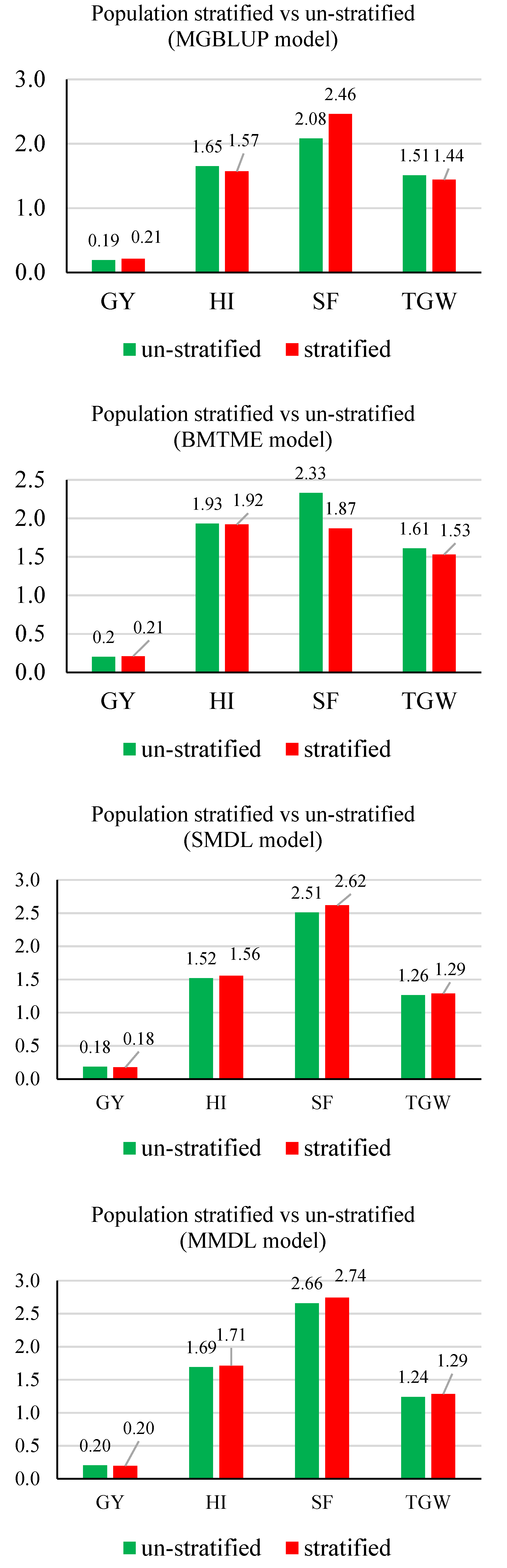
3.2. Prediction Accuracy
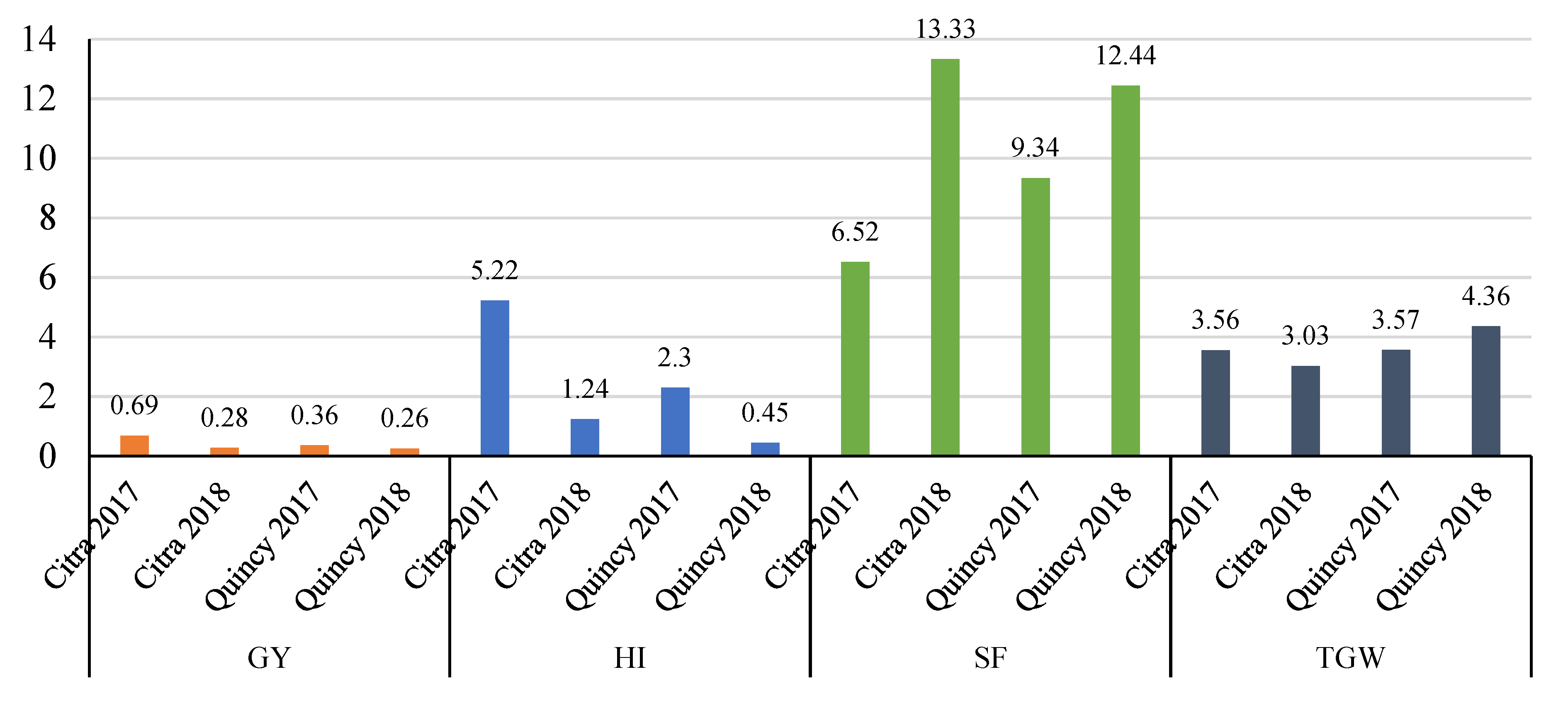
3.3. Response to Selection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mann, J.; Cummings, J.; Englyst, H.; Key, T.; Liu, S.; Riccardi, G.; Summerbell, C.; Uauy, R.; Van Dam, R.; Venn, B. FAO/WHO scientific update on carbohydrates in human nutrition: Conclusions. Eur. J. Clin. Nutr. 2007, 61, S132–S137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, A. Plant Breeding for Water-Limited Environments; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2010; ISBN 1-4419-7491-1. [Google Scholar]
- Ceccarelli, S.; Grando, S.; Maatougui, M.; Michael, M.; Slash, M.; Haghparast, R.; Rahmanian, M.; Taheri, A.; Al-Yassin, A.; Benbelkacem, A. Plant breeding and climate changes. J. Agric. Sci. 2010, 148, 627–637. [Google Scholar] [CrossRef]
- Tester, M.; Langridge, P. Breeding technologies to increase crop production in a changing world. Science 2010, 327, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.E.; Sleeter, B.M.; Abatzoglou, J.T.; Antle, J.M. Climate impacts on agricultural land use in the USA: The role of socio-economic scenarios. Clim. Chang. 2017, 144, 329–345. [Google Scholar] [CrossRef] [Green Version]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of Total Genetic Value Using Genome-Wide Dense Marker Maps. Genetics 2001, 157, 1819. [Google Scholar]
- Marroni, F.; Pinosio, S.; Morgante, M. The quest for rare variants: Pooled multiplexed next generation sequencing in plants. Front. Plant Sci. 2012, 3, 133. [Google Scholar] [CrossRef] [Green Version]
- Bhat, J.A.; Ali, S.; Salgotra, R.K.; Mir, Z.A.; Dutta, S.; Jadon, V.; Tyagi, A.; Mushtaq, M.; Jain, N.; Singh, P.K. Genomic selection in the era of next generation sequencing for complex traits in plant breeding. Front. Genet. 2016, 7, 221. [Google Scholar] [CrossRef] [Green Version]
- Eathington, S.R.; Crosbie, T.M.; Edwards, M.D.; Reiter, R.S.; Bull, J.K. Molecular markers in a commercial breeding program. Crop Sci. 2007, 47, S154–S163. [Google Scholar] [CrossRef]
- Battenfield, S.D.; Guzmán, C.; Gaynor, R.C.; Singh, R.P.; Peña, R.J.; Dreisigacker, S.; Fritz, A.K.; Poland, J.A. Genomic selection for processing and end-use quality traits in the CIMMYT spring bread wheat breeding program. Plant Genome 2016, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B Stat. Methodol. 2005, 67, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Hastie, T.; Tibshirani, R.; Wainwright, M. Statistical Learning with Sparsity: The Lasso and Generalizations; CRC Press: Boca Raton, FL, USA, 2015; ISBN 1-4987-1217-7. [Google Scholar]
- Habier, D.; Fernando, R.L.; Kizilkaya, K.; Garrick, D.J. Extension of the Bayesian alphabet for genomic selection. BMC Bioinform. 2011, 12, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianola, D. Priors in whole-genome regression: The Bayesian alphabet returns. Genetics 2013, 194, 573–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Rodríguez, P.; Crossa, J.; Rutkoski, J.; Poland, J.; Singh, R.; Legarra, A.; Autrique, E.; de Los Campos, G.; Burgueño, J.; Dreisigacker, S. Single-step genomic and pedigree genotype × environment interaction models for predicting wheat lines in international environments. Plant Genome 2017, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Jannink, J.-L. Multiple-trait genomic selection methods increase genetic value prediction accuracy. Genetics 2012, 192, 1513–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyra, D.H.; de Freitas Mendonça, L.; Galli, G.; Alves, F.C.; Granato, Í.S.C.; Fritsche-Neto, R. Multi-trait genomic prediction for nitrogen response indices in tropical maize hybrids. Mol. Breed. 2017, 37, 80. [Google Scholar] [CrossRef]
- Fernandes, S.B.; Dias, K.O.; Ferreira, D.F.; Brown, P.J. Efficiency of multi-trait, indirect, and trait-assisted genomic selection for improvement of biomass sorghum. Theor. Appl. Genet. 2018, 131, 747–755. [Google Scholar] [CrossRef] [Green Version]
- Schulthess, A.W.; Wang, Y.; Miedaner, T.; Wilde, P.; Reif, J.C.; Zhao, Y. Multiple-trait-and selection indices-genomic predictions for grain yield and protein content in rye for feeding purposes. Theor. Appl. Genet. 2016, 129, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.; Panozzo, J.; Walker, C.; Choy, A.; Kant, S.; Wong, D.; Tibbits, J.; Daetwyler, H.; Rochfort, S.; Hayden, M. Accelerating wheat breeding for end-use quality with multi-trait genomic predictions incorporating near infrared and nuclear magnetic resonance-derived phenotypes. Theor. Appl. Genet. 2017, 130, 2505–2519. [Google Scholar] [CrossRef]
- Jiang, J.; Zhang, Q.; Ma, L.; Li, J.; Wang, Z.; Liu, J. Joint prediction of multiple quantitative traits using a Bayesian multivariate antedependence model. Heredity 2015, 115, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgueño, J.; Crossa, J.; Cotes, J.M.; Vicente, F.S.; Das, B. Prediction assessment of linear mixed models for multienvironment trials. Crop Sci. 2011, 51, 944–954. [Google Scholar] [CrossRef]
- Montesinos-López, O.A.; Montesinos-López, A.; Pérez-Rodríguez, P.; de los Campos, G.; Eskridge, K.; Crossa, J. Threshold models for genome-enabled prediction of ordinal categorical traits in plant breeding. G3 Genes Genomes Genet. 2015, 5, 291–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montesinos-López, A.; Montesinos-López, O.A.; Cuevas, J.; Mata-López, W.A.; Burgueño, J.; Mondal, S.; Huerta, J.; Singh, R.; Autrique, E.; González-Pérez, L. Genomic Bayesian functional regression models with interactions for predicting wheat grain yield using hyper-spectral image data. Plant Methods 2017, 13, 62. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Pradhan, S.; Shahi, D.; Khan, J.; Mcbreen, J.; Bai, G.; Murphy, J.P.; Babar, M.A. Increased prediction Accuracy Using combined Genomic information and physiological traits in A Soft Wheat panel evaluated in Multi-environments. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Crain, J.; Mondal, S.; Rutkoski, J.; Singh, R.P.; Poland, J. Combining high-throughput phenotyping and genomic information to increase prediction and selection accuracy in wheat breeding. Plant Genome 2018, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Krause, M.R.; González-Pérez, L.; Crossa, J.; Pérez-Rodríguez, P.; Montesinos-López, O.; Singh, R.P.; Dreisigacker, S.; Poland, J.; Rutkoski, J.; Sorrells, M. Hyperspectral reflectance-derived relationship matrices for genomic prediction of grain yield in wheat. G3 Genes Genomes Genet. 2019, 9, 1231–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montesinos-López, O.A.; Montesinos-López, A.; Crossa, J.; Toledo, F.H.; Pérez-Hernández, O.; Eskridge, K.M.; Rutkoski, J. A genomic Bayesian multi-trait and multi-environment model. G3 Genes Genomes Genet. 2016, 6, 2725–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montesinos-López, O.A.; Montesinos-López, A.; Crossa, J.; Cuevas, J.; Montesinos-López, J.C.; Gutiérrez, Z.S.; Lillemo, M.; Philomin, J.; Singh, R. A Bayesian genomic multi-output regressor stacking model for predicting multi-trait multi-environment plant breeding data. G3 Genes Genomes Genet. 2019, 9, 3381–3393. [Google Scholar] [CrossRef] [Green Version]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Brief. Bioinform. 2017, 18, 851–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, D.R.; Snoek, J.; Rinn, J.L. Basset: Learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 2016, 26, 990–999. [Google Scholar] [CrossRef] [Green Version]
- Bellot, P.; de los Campos, G.; Pérez-Enciso, M. Can deep learning improve genomic prediction of complex human traits? Genetics 2018, 210, 809–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdollahi-Arpanahi, R.; Gianola, D.; Peñagaricano, F. Deep learning versus parametric and ensemble methods for genomic prediction of complex phenotypes. Genet. Sel. Evol. 2020, 52, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, D. Application of Deep Learning in Genomic Selection; IEEE: Piscataway, NJ, USA, 2017; p. 2280. [Google Scholar]
- Ma, W.; Qiu, Z.; Song, J.; Cheng, Q.; Ma, C. DeepGS: Predicting phenotypes from genotypes using Deep Learning. bioRxiv 2017. [Google Scholar] [CrossRef] [Green Version]
- Montesinos-López, O.A.; Montesinos-López, A.; Crossa, J.; Gianola, D.; Hernández-Suárez, C.M.; Martín-Vallejo, J. Multi-trait, multi-environment deep learning modeling for genomic-enabled prediction of plant traits. G3 Genes Genomes Genet. 2018, 8, 3829–3840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jannink, J.-L.; Lorenz, A.J.; Iwata, H. Genomic selection in plant breeding: From theory to practice. Brief. Funct. Genom. 2010, 9, 166–177. [Google Scholar] [CrossRef] [Green Version]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.-L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef] [Green Version]
- Windhausen, V.S.; Atlin, G.N.; Hickey, J.M.; Crossa, J.; Jannink, J.-L.; Sorrells, M.E.; Raman, B.; Cairns, J.E.; Tarekegne, A.; Semagn, K. Effectiveness of genomic prediction of maize hybrid performance in different breeding populations and environments. G3 Genes Genomes Genet. 2012, 2, 1427–1436. [Google Scholar] [CrossRef] [Green Version]
- Isidro, J.; Jannink, J.-L.; Akdemir, D.; Poland, J.; Heslot, N.; Sorrells, M.E. Training set optimization under population structure in genomic selection. Theor. Appl. Genet. 2015, 128, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Arruda, M.; Lipka, A.E.; Brown, P.J.; Krill, A.; Thurber, C.; Brown-Guedira, G.; Dong, Y.; Foresman, B.; Kolb, F.L. Comparing genomic selection and marker-assisted selection for Fusarium head blight resistance in wheat (Triticum aestivum L.). Mol. Breed. 2016, 36, 84. [Google Scholar] [CrossRef]
- Shearman, V.; Sylvester-Bradley, R.; Scott, R.; Foulkes, M. Physiological processes associated with wheat yield progress in the UK. Crop Sci. 2005, 45, 175–185. [Google Scholar]
- Abbate, P.E.; López, J.R.; Brach, A.M.; Gutheim, F.; Gonzalez, F.; Kruk, B.; Serrago, R. Fertilidad de las espigas de trigo en ambientes sub-potenciales. In Workshop Internacional: Ecofisiología Vegetal Aplicada al Estudio de la Determinación del Rendimiento y la Calidad de los Cultivos de Granos, Mar del Plata, Buenos Aires, Argentina, 6–7 September 2007; Kruk, B., Serrago, R., Eds.; FAUBA: Buenos Aires, Argentina, 2007; pp. 2–3. [Google Scholar]
- Abbate, P.E.; Pontaroli, A.C.; Lázaro, L.; Gutheim, F. A method of screening for spike fertility in wheat. J. Agric. Sci. 2013, 151, 322–330. [Google Scholar] [CrossRef]
- Acreche, M.M.; Briceño-Félix, G.; Sánchez, J.A.M.; Slafer, G.A. Physiological bases of genetic gains in Mediterranean bread wheat yield in Spain. Eur. J. Agron. 2008, 28, 162–170. [Google Scholar] [CrossRef]
- Pradhan, S.; Babar, M.A.; Robbins, K.; Bai, G.; Mason, R.E.; Khan, J.; Shahi, D.; Avci, M.; Guo, J.; Hossain, M.M. Understanding the Genetic Basis of Spike Fertility to Improve Grain Number, Harvest Index, and Grain Yield in Wheat Under High Temperature Stress Environments. Front. Plant Sci. 2019, 10, 1481. [Google Scholar] [CrossRef] [Green Version]
- Botwright, T.L.; Condon, A.G.; Rebetzke, G.J.; Richards, R.A. Field evaluation of early vigour for genetic improvement of grain yield in wheat. Aust. J. Agric. Res. 2002, 53, 1137–1145. [Google Scholar] [CrossRef]
- Kuchel, H.; Williams, K.J.; Langridge, P.; Eagles, H.A.; Jefferies, S.P. Genetic dissection of grain yield in bread wheat. I. QTL analysis. Theor. Appl. Genet. 2007, 115, 1029–1041. [Google Scholar] [CrossRef]
- Reynolds, M.; Foulkes, M.J.; Slafer, G.A.; Berry, P.; Parry, M.A.; Snape, J.W.; Angus, W.J. Raising yield potential in wheat. J. Exp. Bot. 2009, 60, 1899–1918. [Google Scholar] [CrossRef] [Green Version]
- Fischer, R. Wheat physiology: A review of recent developments. Crop Pasture Sci. 2011, 62, 95–114. [Google Scholar] [CrossRef] [Green Version]
- Parry, M.; Slafer, G. Achieving yield gains in wheat. Plant Cell Environ. 2012, 35, 17991823Sears. [Google Scholar]
- Gaju, O.; Reynolds, M.P.; Sparkes, D.L.; Foulkes, M.J. Relationships between Large-Spike Phenotype, Grain Number, and Yield Potential in Spring Wheat. Crop Sci. 2009, 49, 961–973. [Google Scholar] [CrossRef]
- González, F.G.; Terrile, I.I.; Falcón, M.O. Spike Fertility and Duration of Stem Elongation as Promising Traits to Improve Potential Grain Number (and Yield): Variation in Modern Argentinean Wheats. Crop Sci. 2011, 51, 1693–1702. [Google Scholar] [CrossRef]
- Rivera-Amado, C.; Trujillo-Negrellos, E.; Sylvester-Bradley, R.; Molero, G.; Sierra-Gonzalez, A.; Reynolds, M.; Foulkes, J. Achieving increases in spike growth, fruiting efficiency, and harvest index in high biomass wheat cultivars. In Proceedings of the 2nd International TRIGO (Wheat) Yield Potential; CIMMYT: Mexico City, Mexico, 2016; p. 70. [Google Scholar]
- Molero, G.; Joynson, R.; Pinera-Chavez, F.J.; Gardiner, L.; Rivera-Amado, C.; Hall, A.; Reynolds, M.P. Elucidating the genetic basis of biomass accumulation and radiation use efficiency in spring wheat and its role in yield potential. Plant Biotechnol. J. 2019, 17, 1276–1288. [Google Scholar] [CrossRef]
- Lopes, M.S.; El-Basyoni, I.; Baenziger, P.S.; Singh, S.; Royo, C.; Ozbek, K.; Aktas, H.; Ozer, E.; Ozdemir, F.; Manickavelu, A.; et al. Exploiting genetic diversity from landraces in wheat breeding for adaptation to climate change. J. Exp. Bot. 2015, 66, 3477–3486. [Google Scholar] [CrossRef]
- Martino, D.L.; Abbate, P.E.; Cendoya, M.G.; Gutheim, F.; Mirabella, N.E.; Pontaroli, A.C. Wheat spike fertility: Inheritance and relationship with spike yield components in early generations. Plant Breed. 2015, 134, 264–270. [Google Scholar] [CrossRef]
- Thavamanikumar, S.; Dolferus, R.; Thumma, B.R. Comparison of Genomic Selection Models to Predict Flowering Time and Spike Grain Number in Two Hexaploid Wheat Doubled Haploid Populations. G3 Genes Genomes Genet. 2015, 5, 1991. [Google Scholar] [CrossRef] [Green Version]
- Federer, W.T.; Raghavarao, D. On augmented designs. Biometrics 1975, 31, 29–35. [Google Scholar] [CrossRef] [Green Version]
- Zadoks, J.C.; Chang, T.T.; Konzak, C.F. A decimal code for the growth stages of cereals. Weed Res. 1974, 14, 415–421. [Google Scholar] [CrossRef]
- Littell, R.C.; Milliken, G.A.; Stroup, W.W.; Wolfinger, R.D. SAS system for mixed models. Technometrics 1997, 39, 344. [Google Scholar]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Poland, J.; Endelman, J.; Dawson, J.; Rutkoski, J.; Wu, S.; Manes, Y.; Dreisigacker, S.; Crossa, J.; Sánchez-Villeda, H.; Sorrells, M. Genomic selection in wheat breeding using genotyping-by-sequencing. Plant Genome 2012, 5, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Bansal, V.; Harismendy, O.; Tewhey, R.; Murray, S.S.; Schork, N.J.; Topol, E.J.; Frazer, K.A. Accurate detection and genotyping of SNPs utilizing population sequencing data. Genome Res. 2010, 20, 537–545. [Google Scholar] [CrossRef] [Green Version]
- Montesinos-López, O.A.; Montesinos-López, A.; Luna-Vázquez, F.J.; Toledo, F.H.; Pérez-Rodríguez, P.; Lillemo, M.; Crossa, J. An R package for Bayesian analysis of multi-environment and multi-trait multi-environment data for genome-based prediction. G3 Genes Genomes Genet. 2019, 9, 1355–1369. [Google Scholar] [CrossRef] [Green Version]
- Spyromitros-Xioufis, E.; Tsoumakas, G.; Groves, W.; Vlahavas, I. Multi-label classification methods for multi-target regression. arXiv 2012, arXiv:12116581. [Google Scholar]
- Spyromitros-Xioufis, E.; Tsoumakas, G.; Groves, W.; Vlahavas, I. Multi-target regression via input space expansion: Treating targets as inputs. Mach. Learn. 2016, 104, 55–98. [Google Scholar] [CrossRef] [Green Version]
- Džeroski, S.; Demšar, D.; Grbović, J. Predicting chemical parameters of river water quality from bioindicator data. Appl. Intell. 2000, 13, 7–17. [Google Scholar] [CrossRef]
- Kocev, D.; Džeroski, S.; White, M.D.; Newell, G.R.; Griffioen, P. Using single-and multi-target regression trees and ensembles to model a compound index of vegetation condition. Ecol. Model. 2009, 220, 1159–1168. [Google Scholar] [CrossRef]
- Angermueller, C.; Lee, H.J.; Reik, W.; Stegle, O. DeepCpG: Accurate prediction of single-cell DNA methylation states using deep learning. Genome Biol. 2017, 18, 67. [Google Scholar] [CrossRef] [Green Version]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [Green Version]
- Falconer, D.S.; Mackay, T.F.; Frankham, R. Introduction to quantitative genetics (4th edn). Trends Genet. 1996, 12, 280. [Google Scholar]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting linear mixed-effects models using lme4. arXiv 2014, arXiv:14065823. [Google Scholar]
- Pérez, P.; de Los Campos, G. Genome-wide regression and prediction with the BGLR statistical package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef]
- Abadi, M.; Barham, P.; Chen, J.; Chen, Z.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Irving, G.; Isard, M. Tensorflow: A System for Large-Scale Machine Learning. In Proceedings of the 12th USENIX Symposium on Operating Systems Design and Implementation (OSDI ’16), Savannah, GA, USA, 2–4 November 2016; pp. 265–283. [Google Scholar]
- Gulli, A.; Pal, S. Deep Learning with KERAS; Packt Publishing Ltd.: Birmingham, UK, 2017; ISBN 1-78712-903-9. [Google Scholar]
- Lenth, R.V. Response-surface methods in R, using rsm. J. Stat. Softw. 2009, 32, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Banta, S.J. Symposium on Potential Productivity of Field Crops under Different Environments; International Rice Research Institute: Los Banos, Philippine, 1983; Volume 15, ISBN 971-10-4114-6. [Google Scholar]
- Foulkes, M.J.; Slafer, G.A.; Davies, W.J.; Berry, P.M.; Sylvester-Bradley, R.; Martre, P.; Calderini, D.F.; Griffiths, S.; Reynolds, M.P. Raising yield potential of wheat. III. Optimizing partitioning to grain while maintaining lodging resistance. J. Exp. Bot. 2011, 62, 469–486. [Google Scholar] [CrossRef] [Green Version]
- Slafer, G.A.; Elia, M.; Savin, R.; García, G.A.; Terrile, I.I.; Ferrante, A.; Miralles, D.J.; González, F.G. Fruiting efficiency: An alternative trait to further rise wheat yield. Food Energy Secur. 2015, 4, 92–109. [Google Scholar] [CrossRef]
- Ward, B.P.; Brown-Guedira, G.; Tyagi, P.; Kolb, F.L.; Van Sanford, D.A.; Sneller, C.H.; Griffey, C.A. Multienvironment and multitrait genomic selection models in unbalanced early-generation wheat yield trials. Crop Sci. 2019, 59, 491–507. [Google Scholar] [CrossRef] [Green Version]
- Rutkoski, J.; Poland, J.; Mondal, S.; Autrique, E.; Pérez, L.G.; Crossa, J.; Reynolds, M.; Singh, R. Canopy temperature and vegetation indices from high-throughput phenotyping improve accuracy of pedigree and genomic selection for grain yield in wheat. G3 Genes Genomes Genet. 2016, 6, 2799–2808. [Google Scholar] [CrossRef] [Green Version]
- Montesinos-López, O.A.; Montesinos-López, A.; Montesinos-López, J.C.; Crossa, J.; Luna-Vázquez, F.J.; Salinas-Ruiz, J. A Bayesian Multiple-Trait and Multiple-Environment Model Using the Matrix Normal Distribution. Phys. Methods Stimul. Plant Mushroom Dev. 2018, 19. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Zhang, N.; Wang, Y.-G.; George, A.W.; Reverter, A.; Li, Y. Genomic prediction of breeding values using a subset of SNPs identified by three machine learning methods. Front. Genet. 2018, 9, 237. [Google Scholar] [CrossRef]
- Heslot, N.; Jannink, J.-L.; Sorrells, M.E. Using genomic prediction to characterize environments and optimize prediction accuracy in applied breeding data. Crop Sci. 2013, 53, 921–933. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | Year | Coordinates | Soil Type 1 |
|---|---|---|---|
| Citra | 2016–2017 | 29°24′18″ N 82°10′22″ W | Well-drained sandy soil with loamy subsoil at 20–80 inches |
| 2017–2018 | 29°24′32″ N 82°10′46′’ W | ||
| Quincy | 2016–2017 | 30°33′04″ N 84°35′51″ W | Well-drained loamy soils |
| 2017–2018 | 30°32′45″ N 84°35′46″ W |
| Trait | BLUE | SE | H2 | CV | Min | Max | |
|---|---|---|---|---|---|---|---|
| Citra 2017 | GY | 2.0 | 0.1 | 0.71 | 28.3 | 0.3 | 4.5 |
| HI | 30.5 | 0.8 | 0.78 | 17.8 | 16 | 52 | |
| SF | 63.9 | 1.7 | 0.38 | 27.2 | 12 | 142 | |
| TGW | 34.7 | 0.4 | 0.48 | 10.8 | 24 | 48 | |
| Citra 2018 | GY | 3.8 | 0.1 | 0.80 | 11.5 | 1.0 | 7.0 |
| HI | 37.4 | 0.4 | 0.74 | 6.7 | 20 | 48 | |
| SF | 98.3 | 1.2 | 0.68 | 9.5 | 62 | 161 | |
| TGW | 34.1 | 0.4 | 0.87 | 5.1 | 19 | 46 | |
| Quincy 2017 | GY | 3.3 | 0.1 | 0.36 | 16.6 | 1.5 | 5.6 |
| HI | 34.3 | 0.4 | 0.43 | 12.6 | 20 | 47 | |
| SF | 83.2 | 1.7 | 0.22 | 25.1 | 34 | 148 | |
| TGW | 39.4 | 0.3 | 0.58 | 7.4 | 26 | 50 | |
| Quincy 2018 | GY | 5.3 | 0.1 | 0.24 | 18.4 | 2.1 | 8.8 |
| HI | 42.7 | 0.3 | 0.26 | 9.8 | 28 | 54 | |
| SF | 94.6 | 1.4 | 0.32 | 15.6 | 52 | 158 | |
| TGW | 40.9 | 0.4 | 0.44 | 9.7 | 30 | 54 |
| GY | HI | SF | TGW | |
|---|---|---|---|---|
| GY | 0.67 | 0.17 | 0.18 | |
| HI | 0.76 | 0.17 | 0.10 | |
| SF | 0.36 | 0.30 | −0.32 | |
| TGW | 0.33 | 0.24 | −0.23 |
| Quincy 2017 | Citra 2017 | Quincy 2018 | Citra 2018 | |
|---|---|---|---|---|
| Quincy 2017 | 0.24 | 0.26 | 0.19 | |
| Citra 2017 | 0.19 | 0.17 | ||
| Quincy 2018 | 0.16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, J.; Khan, J.; Pradhan, S.; Shahi, D.; Khan, N.; Avci, M.; Mcbreen, J.; Harrison, S.; Brown-Guedira, G.; Murphy, J.P.; et al. Multi-Trait Genomic Prediction of Yield-Related Traits in US Soft Wheat under Variable Water Regimes. Genes 2020, 11, 1270. https://doi.org/10.3390/genes11111270
Guo J, Khan J, Pradhan S, Shahi D, Khan N, Avci M, Mcbreen J, Harrison S, Brown-Guedira G, Murphy JP, et al. Multi-Trait Genomic Prediction of Yield-Related Traits in US Soft Wheat under Variable Water Regimes. Genes. 2020; 11(11):1270. https://doi.org/10.3390/genes11111270
Chicago/Turabian StyleGuo, Jia, Jahangir Khan, Sumit Pradhan, Dipendra Shahi, Naeem Khan, Muhsin Avci, Jordan Mcbreen, Stephen Harrison, Gina Brown-Guedira, Joseph Paul Murphy, and et al. 2020. "Multi-Trait Genomic Prediction of Yield-Related Traits in US Soft Wheat under Variable Water Regimes" Genes 11, no. 11: 1270. https://doi.org/10.3390/genes11111270