The Diversity of Genetic Outcomes from CRISPR/Cas Gene Editing is Regulated by the Length of the Symmetrical Donor DNA Template



Abstract

1. Introduction
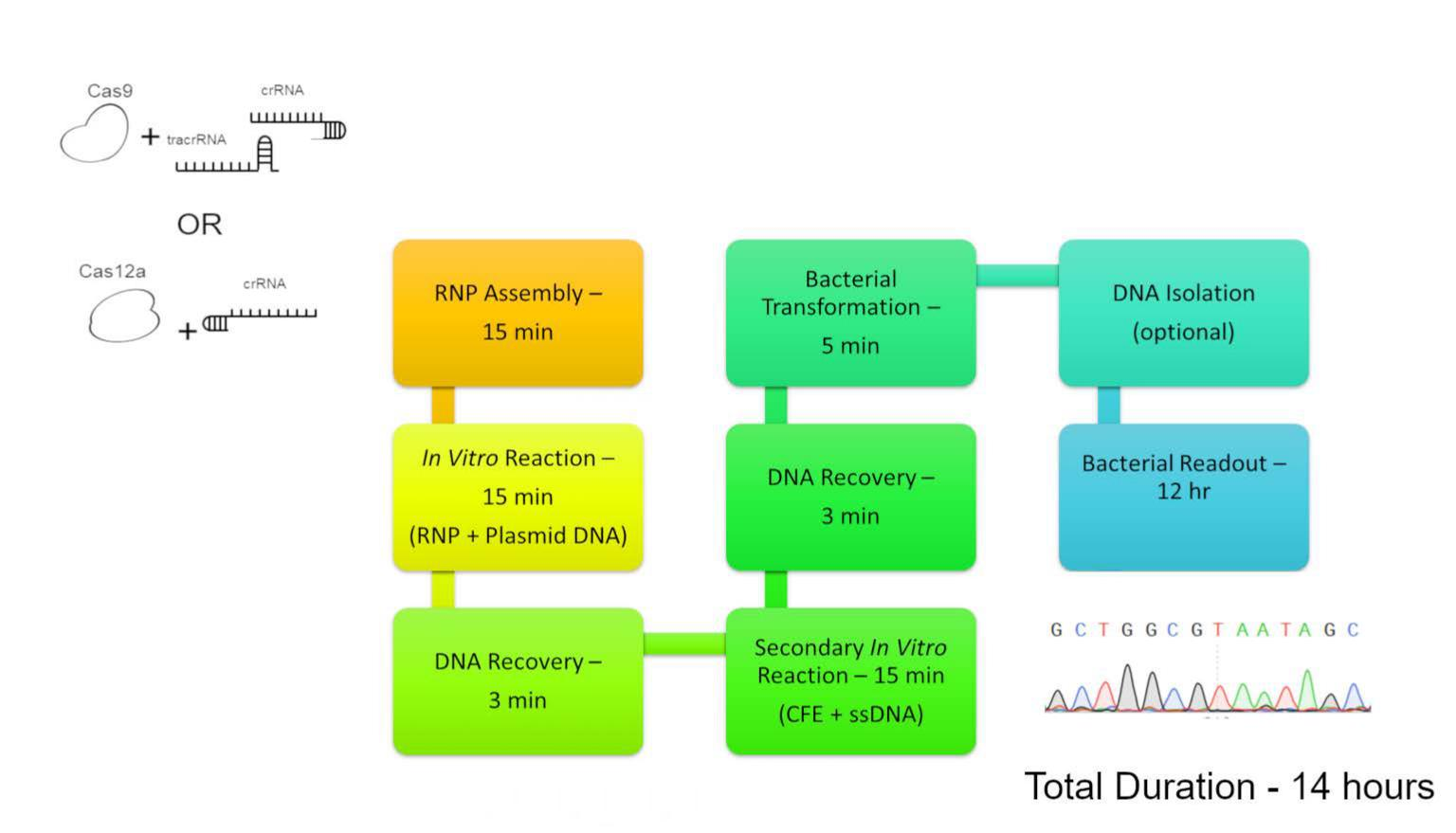
2. Materials and Methods
2.1. Preparing Cell-Free Extract
2.2. Reaction Conditions
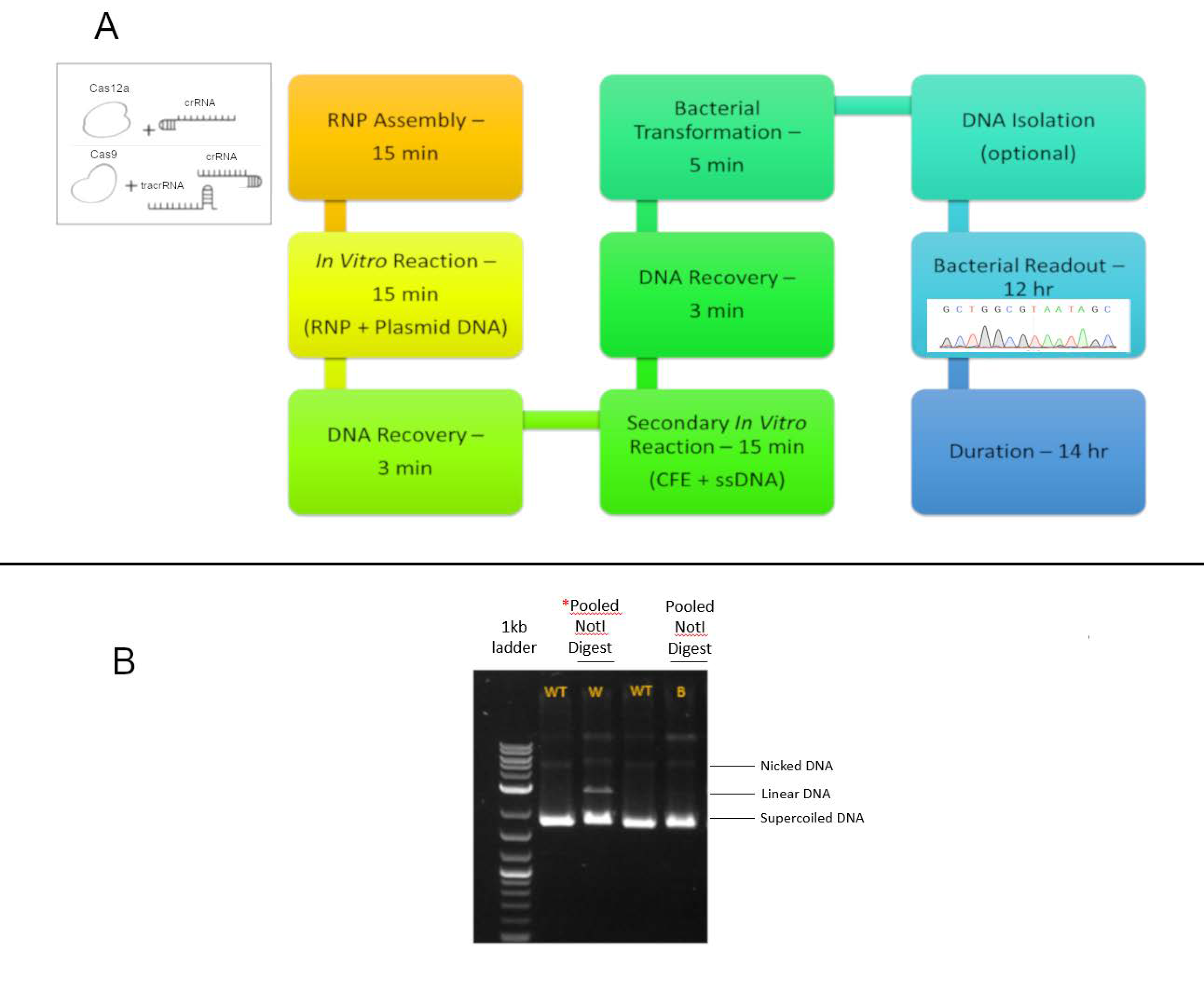
2.3. Transformation, Selection, DNA Isolation, PCR, and Analysis
2.4. Statistical Analysis–Fisher’s Exact Test p < 0.05
3. Results
3.1. Target Site in Genetic Readout System
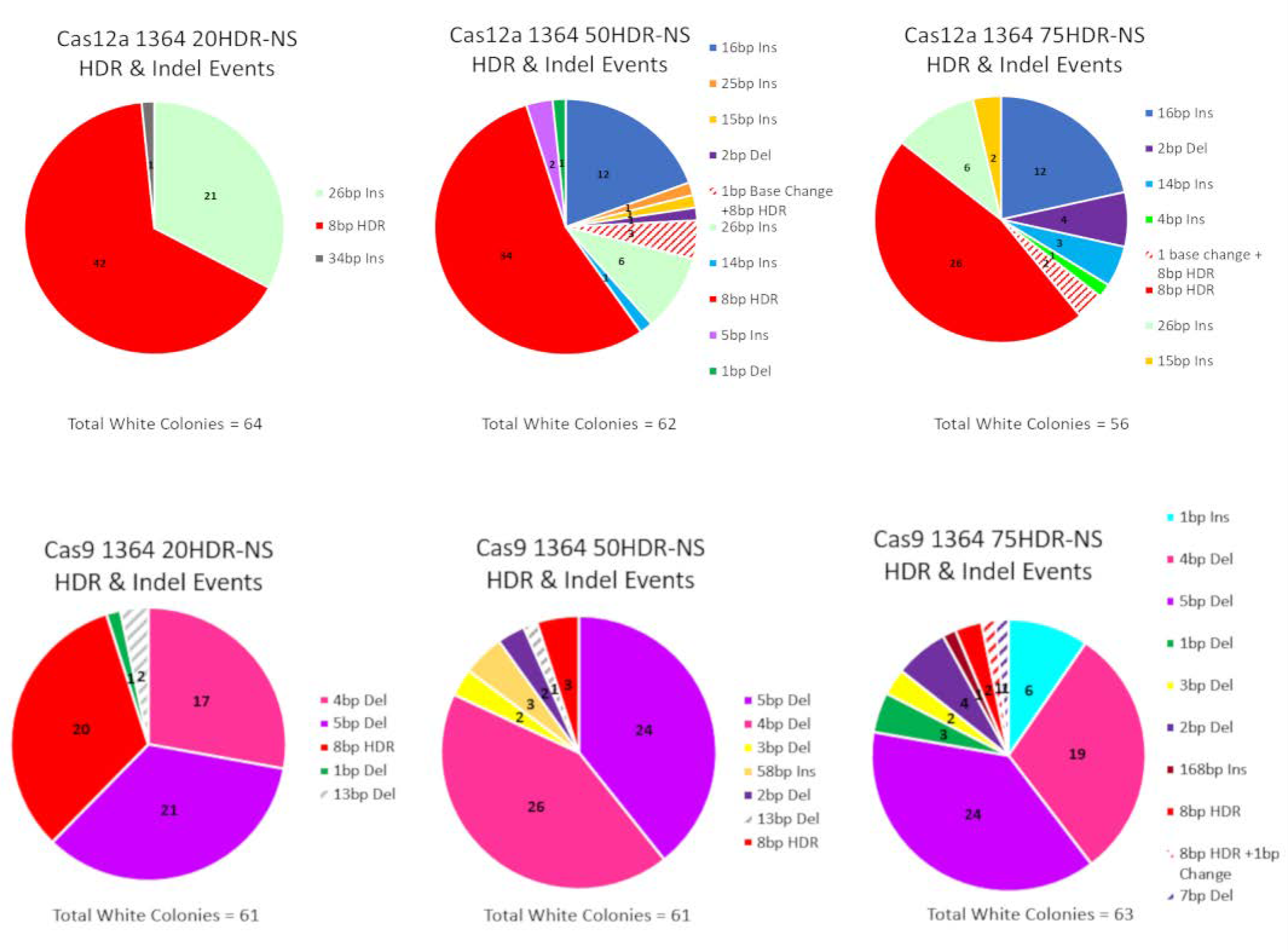
3.1.1. Precise HDR is Influenced by the Length of the Homology Arms of the Donor DNA Template in a Cas12a-Catalyzed Reaction
3.1.2. Precise HDR is Influenced by the Length of the Homology Arms of the Donor DNA Template in a Cas9-Catalyzed Reaction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barrangou, R.; Doudna, J.A. Applications of CRISPR technologies in research and beyond. Nat. Biotechnol. 2016, 34, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 1234–1257. [Google Scholar] [CrossRef] [PubMed]
- Prakash, V.; Moore, M.; Yáñez-Muñoz, R.J. Current progress in therapeutic gene editing for monogenic diseases. Mol. Ther. 2016, 24, 465–474. [Google Scholar] [CrossRef]
- Lessard, S.; Francioli, L.; Alfoldi, J.; Tardif, J.-C.; Ellinor, P.T.; MacArthur, D.G.; Lettre, G.; Orkin, S.H.; Canver, M.C. Human genetic variation alters CRISPR-Cas9 on- and off-targeting specificity at therapeutically implicated loci. Proc. Natl. Acad. Sci. USA 2017, 114, E11257–E11266. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, K.I.; Sutrisnoh, N.A.B.; Srinivasan, H.; Zhang, J.; Li, J.; Zhang, F.; Lalith, C.R.J.; Xing, H.; Shanmugam, R.; et al. Systematic evaluation of CRISPR-Cas systems reveals design principles for genome editing in human cells. Genome Biol. 2018, 19, 1–16. [Google Scholar] [CrossRef]
- Liu, J.; Majumdar, A.; Liu, J.; Thompson, L.H.; Seidman, M.M. Sequence conversion by single strand oligonucleotide donors via non-homologous end joining in mammalian cells. J. Biol. Chem. 2010, 285, 23198–23207. [Google Scholar] [CrossRef]
- Jasin, M.; Haber, J.E. The democratization of gene editing: Insights from site-specific cleavage and double-strand break repair. DNA Repair 2016, 44, 6–16. [Google Scholar] [CrossRef]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 2014, 3, e04766. [Google Scholar] [CrossRef]
- Engstrom, J.U.; Suzuki, T.; Kmiec, E.B. Regulation of targeted gene repair by intrinsic cellular processes. BioEssays 2009, 31, 159–168. [Google Scholar] [CrossRef]
- Engstrom, J.U.; Kmiec, E.B. DNA Replication, cell cycle progression and the targeted gene repair reaction. Cell Cycle 2008, 7, 1402–1414. [Google Scholar] [CrossRef]
- Strouse, B.; Bialk, P.; Niamat, R.A.; Rivera-Torres, N.; Kmiec, E.B. Combinatorial gene editing in mammalian cells using SsODNs and TALENs. Sci. Rep. 2015, 4, 3791. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Torres, N.; Strouse, B.; Bialk, P.; Niamat, R.A.; Kmiec, E.B. The position of DNA cleavage by TALENs and cell synchronization influences the frequency of gene editing directed by single-stranded oligonucleotides. PLoS ONE 2014, 9, e96483. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Torres, N.; Banas, K.; Bialk, P.; Bloh, K.M.; Kmiec, E.B. Insertional mutagenesis by CRISPR/Cas9 ribonucleoprotein gene editing in cells targeted for point mutation repair directed by short single-stranded DNA oligonucleotides. PLoS ONE 2017, 12, e0169350. [Google Scholar] [CrossRef] [PubMed]
- Bialk, P.; Rivera-Torres, N.; Strouse, B.; Kmiec, E.B. Regulation of gene editing activity directed by single-stranded oligonucleotides and CRISPR/Cas9 systems. PLoS ONE 2015, 10, e0129308. [Google Scholar] [CrossRef] [PubMed]
- Sansbury, B.M.; Hewes, A.M.; Kmiec, E.B. Understanding the diversity of genetic outcomes from CRISPR-Cas generated homology-directed repair. Commun. Biol. 2019, 2, 458. [Google Scholar] [CrossRef]
- Paquet, D.; Kwart, D.; Chen, A.; Sproul, A.; Jacob, S.; Teo, S.; Olsen, K.M.; Gregg, A.; Noggle, S.; Tessier-Lavigne, M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 2016, 533, 125–129. [Google Scholar] [CrossRef]
- Kim, D.; Kim, J.; Hur, J.K.; Been, K.W.; Yoon, S.H.; Kim, J.S. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat. Biotechnol. 2016, 34, 863–868. [Google Scholar] [CrossRef]
- Paix, A.; Folkmann, A.; Goldman, D.H.; Kulaga, H.; Grzelak, M.J.; Rasoloson, D.; Paidemarry, S.; Green, R.; Reed, R.R.; Seydoux, G. Precision genome editing using synthesis-dependent repair of Cas9-induced DNA breaks. Proc. Natl. Acad. Sci. USA 2017, 114, E10745–E10754. [Google Scholar] [CrossRef]
- Richardson, C.D.; Kazane, K.R.; Feng, S.J.; Zelin, E.; Bray, N.L.; Schäfer, A.J.; Floor, S.N.; Corn, J.E. CRISPR–Cas9 genome editing in human cells occurs via the fanconi anemia pathway. Nat. Genet. 2018, 50, 1132–1139. [Google Scholar] [CrossRef]
- Brachman, E.E.; Kmiec, E.B. The “biased” evolution of targeted gene repair. Curr. Opin. Mol. Ther. 2002, 4, 171–176. [Google Scholar]
- Jiang, F.; Doudna, J.A. CRISPR–Cas9 structures and mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef]
- Schumann, K.; Lin, S.; Boyer, E.; Simeonov, D.R.; Subramaniam, M.; Gate, R.E.; Haliburton, G.E.; Ye, C.J.; Bluestone, J.A.; Doudna, J.A.; et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc. Natl. Acad. Sci. USA 2015, 112, 10437–10442. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.D.; Ray, G.J.; DeWitt, M.A.; Curie, G.L.; Corn, J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Guell, M.; Byrne, S.; Yang, J.L.; Angeles, A.D.L.; Mali, P.; Aach, J.; Kim-Kiselak, C.; Briggs, A.W.; Rios, X.; et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 2013, 41, 9049–9061. [Google Scholar] [CrossRef] [PubMed]
- Quadros, R.M.; Miura, H.; Harms, D.W.; Akatsuka, H.; Sato, T.; Aida, T.; Redder, R.; Richardson, G.P.; Inagaki, Y.; Sakai, D.; et al. Easi-CRISPR: A robust method for one-step generation of mice carrying conditional and insertion alleles using long SsDNA donors and CRISPR ribonucleoproteins. Genome Biol. 2017, 18, 92. [Google Scholar] [CrossRef]
- Sansbury, B.M.; Wagner, A.M.; Nitzan, E.; Tarcic, G.; Kmiec, E.B. CRISPR-directed In Vitro gene editing of plasmid DNA catalyzed by Cpf1 (Cas12a) nuclease and a mammalian cell-free extract. Cris. J. 2018, 1, 191–202. [Google Scholar] [CrossRef]
- Sansbury, B.M.; Wagner, A.M.; Tarcic, G.; Barth, S.; Nitzan, E.; Goldfus, R.; Vidne, M.; Kmiec, E.B. CRISPR-directed gene editing catalyzes precise gene segment replacement In Vitro enabling a novel method for multiplex site-directed mutagenesis. Cris. J. 2019, 2, 121–132. [Google Scholar] [CrossRef]
- Hewes, A.M.; Sansbury, B.M.; Barth, S.; Tarcic, G.; Kmiec, E.B. GRNA sequence heterology tolerance catalyzed by CRISPR/Cas in an In Vitro homology-directed repair reaction. Mol. Ther. Nucleic Acids 2020, 20, 568–579. [Google Scholar] [CrossRef]
- Boel, A.; Steyaert, W.; De Rocker, N.; Menten, B.; Callewaert, B.; De Paepe, A.; Coucke, P.; Willaert, A. BATCH-GE: Batch analysis of next-generation sequencing data for genome editing assessment. Sci. Rep. 2016, 6, 30330. [Google Scholar] [CrossRef]
- Gratz, S.J.; Cummings, A.M.; Nguyen, J.N.; Hamm, D.C.; Donohue, L.K.; Harrison, M.M.; Wildonger, J.; O’Connor-Giles, K.M. Genome engineering of drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics 2013, 194, 1029–1035. [Google Scholar] [CrossRef]
- Hendel, A.; Bak, R.O.; Clark, J.T.; Kennedy, A.B.; Ryan, D.E.; Roy, S.; Steinfeld, I.; Lunstad, B.D.; Kaiser, R.J.; Wilkens, A.B.; et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015, 33, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Cole-Strauss, A.; Gamper, H.; Holloman, W.K.; Muñoz, M.; Cheng, N.; Kmiec, E.B. Targeted gene repair directed by the chimeric RNA/DNA oligonucleotide in a mammalian cell-free extract. Nucleic Acids Res. 1999, 27, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Boel, A.; De Saffel, H.; Steyaert, W.; Callewaert, B.; De Paepe, A.; Coucke, P.J.; Willaert, A. CRISPR/Cas9-mediated homology-directed repair by SsODNs in zebrafish induces complex mutational patterns resulting from genomic integration of repair-template fragments. Dis. Model. Mech. 2018, 11, dmm035352. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, E.K.; Chen, T.; Amendola, M.; Van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef]
- Connelly, J.P.; Pruett-Miller, S.M. CRIS.Py: A versatile and high-throughput analysis program for CRISPR-based genome editing. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Tsai, S.Q.; Prew, M.S.; Nguyen, N.T.; Welch, M.M.; Lopez, J.M.; Mccaw, Z.R.; Aryee, M.J.; Joung, J.K.; Conceived, J.K.J.; et al. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells HHS public access author manuscript. Nat. Biotechnol. 2016, 34, 869–874. [Google Scholar] [CrossRef]
- Storici, F.; Snipe, J.R.; Chan, G.K.; Gordenin, D.A.; Resnick, M.A. Conservative repair of a chromosomal double-strand break by single-strand DNA through two steps of annealing. Mol. Cell. Biol. 2006, 26, 7645–7657. [Google Scholar] [CrossRef]
- Majumdar, A.; Muniandy, P.A.; Liu, J.; Liu, J.L.; Liu, S.T.; Cuenoud, B.; Seidman, M.M. Targeted gene knock in and sequence modulation mediated by a psoralen-linked triplex-forming oligonucleotide. J. Biol. Chem. 2008, 283, 11244–11252. [Google Scholar] [CrossRef]
- Helleday, T.; Lo, J.; van Gent, D.C.; Engelward, B.P. DNA double-strand break repair: From mechanistic understanding to cancer treatment. DNA Repair 2007, 6, 923–935. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| * Cas12a vs. Cas9-20HDR-NS | ||||
| HDR | Indel | Total | ||
| Cas12a 1364 20HDR-NS | 42 | 22 | 64 | |
| Cas9 1364 20HDR-NS | 20 | 41 | 61 | |
| Marginal column totals | 62 | 63 | 125 | |
| p-value < 0.05 | 0.0003 * | |||
| * Cas12a vs. Cas9 50HDR-NS | ||||
| HDR | Indel | Total | ||
| Cas12a 1364 50HDR-NS | 34 | 28 | 62 | |
| Cas9 1364 50HDR-NS | 3 | 58 | 61 | |
| Marginal column totals | 37 | 86 | 123 | |
| p-value < 0.05 | 0.0001 * | |||
| * Cas12a vs. Cas9-75HDR-NS | ||||
| HDR | Indel | Total | ||
| Cas12a 1364 75HDR-NS | 26 | 30 | 56 | |
| Cas9 1364 75HDR-NS | 2 | 61 | 63 | |
| Marginal column totals | 28 | 91 | 119 | |
| p-value < 0.05 | 0.00001 * | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hewes, A.M.; Sansbury, B.M.; Kmiec, E.B. The Diversity of Genetic Outcomes from CRISPR/Cas Gene Editing is Regulated by the Length of the Symmetrical Donor DNA Template. Genes 2020, 11, 1160. https://doi.org/10.3390/genes11101160
Hewes AM, Sansbury BM, Kmiec EB. The Diversity of Genetic Outcomes from CRISPR/Cas Gene Editing is Regulated by the Length of the Symmetrical Donor DNA Template. Genes. 2020; 11(10):1160. https://doi.org/10.3390/genes11101160
Chicago/Turabian StyleHewes, Amanda M., Brett M. Sansbury, and Eric B. Kmiec. 2020. "The Diversity of Genetic Outcomes from CRISPR/Cas Gene Editing is Regulated by the Length of the Symmetrical Donor DNA Template" Genes 11, no. 10: 1160. https://doi.org/10.3390/genes11101160
APA StyleHewes, A. M., Sansbury, B. M., & Kmiec, E. B. (2020). The Diversity of Genetic Outcomes from CRISPR/Cas Gene Editing is Regulated by the Length of the Symmetrical Donor DNA Template. Genes, 11(10), 1160. https://doi.org/10.3390/genes11101160