The Transcriptomic Landscape of Cupriavidus metallidurans CH34 Acutely Exposed to Copper



Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Media, and Culture Conditions
2.2. RNA Extraction
2.3. TagRNA-Sequencing Protocol
2.4. Differential Gene Expression Calculation and Functional Enrichment Analysis
2.5. Transcriptional Start Site Profiling
2.6. 5′ and 3′ RACE Protocol
2.7. Statistics
3. Results and Discussion
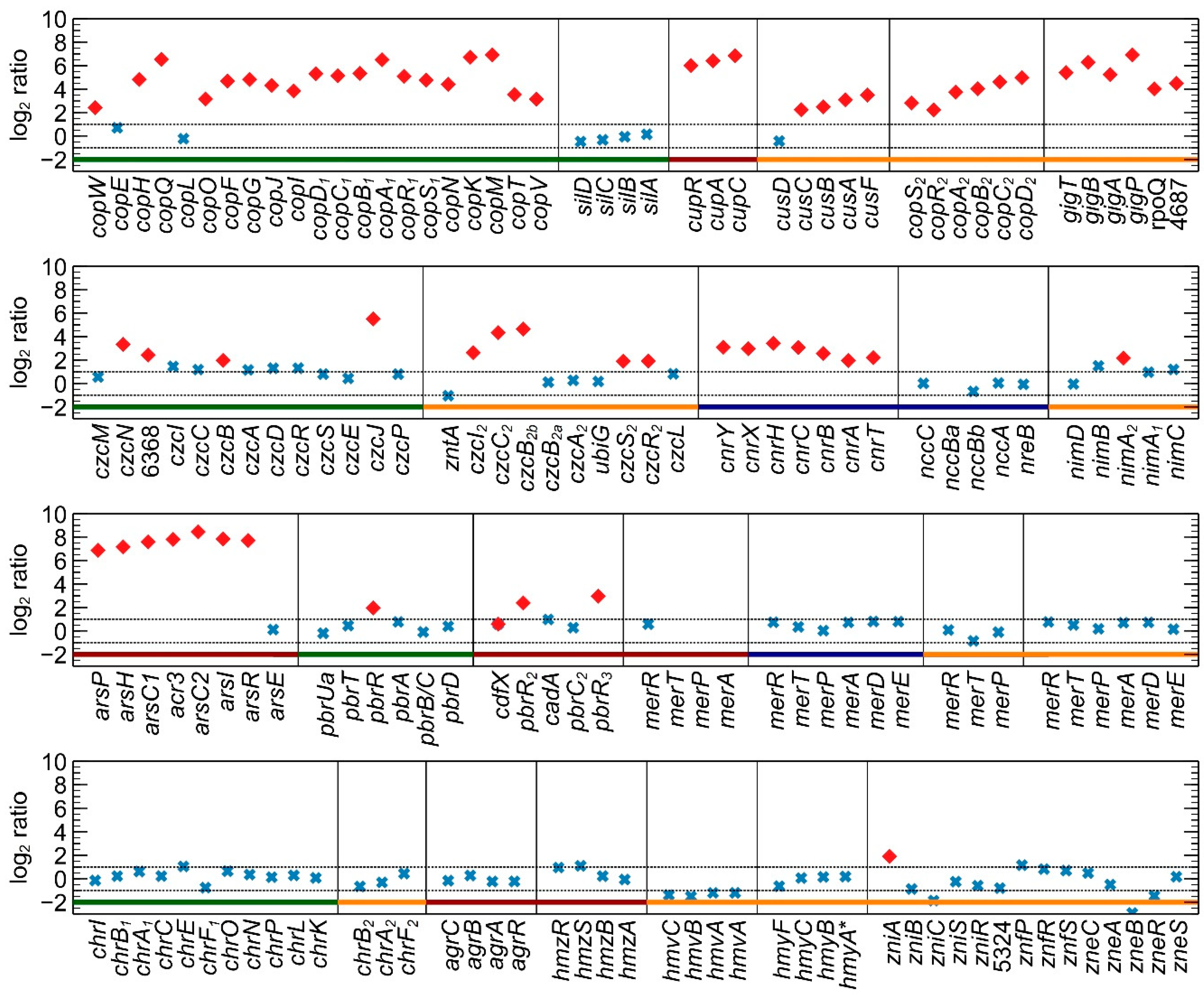
3.1. Differential Gene Expression Analysis
3.2. Transcriptional Start Site Profiling
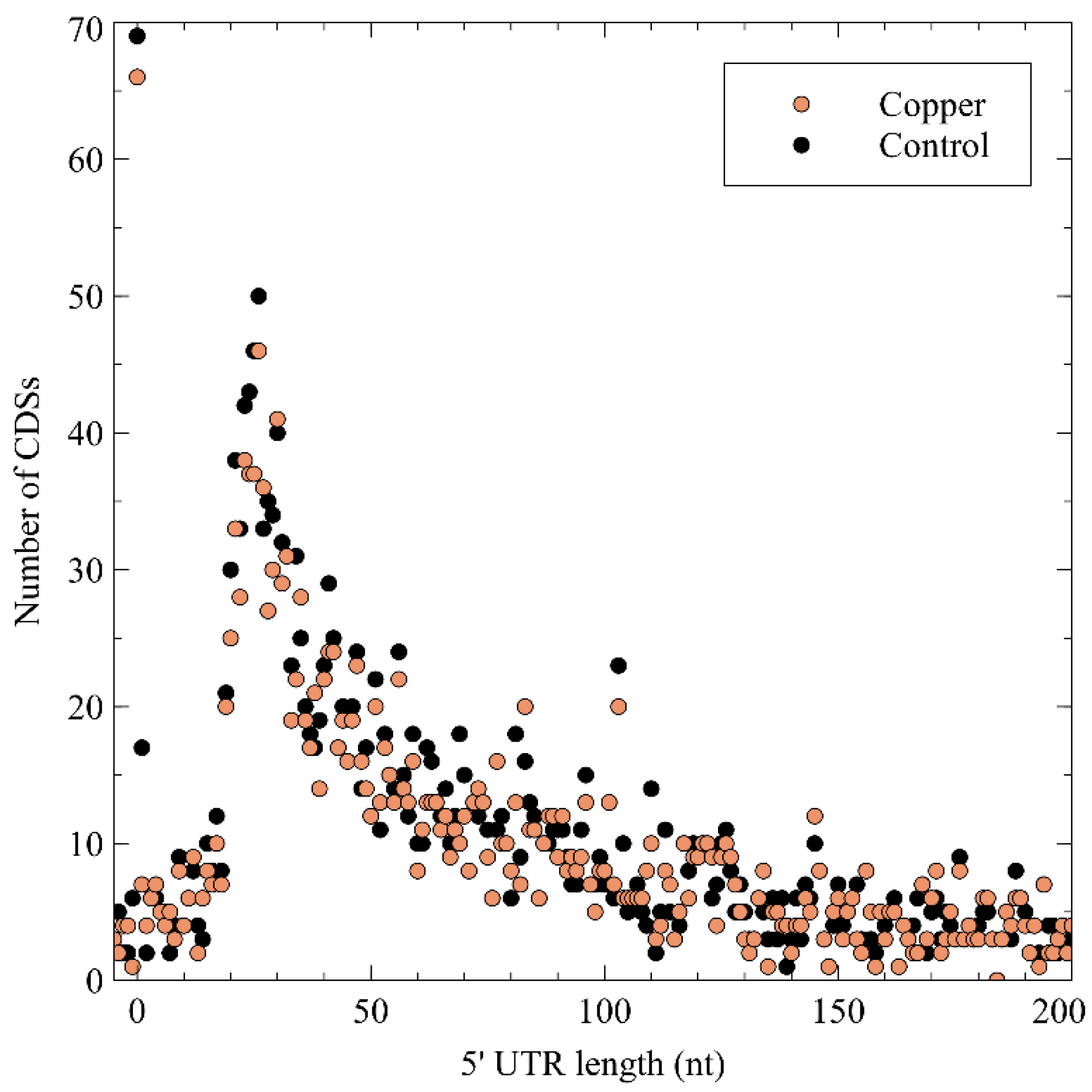
3.2.1. General Characteristics of Detected TSSs
3.2.2. Primary TSSs
3.2.3. Secondary TSSs
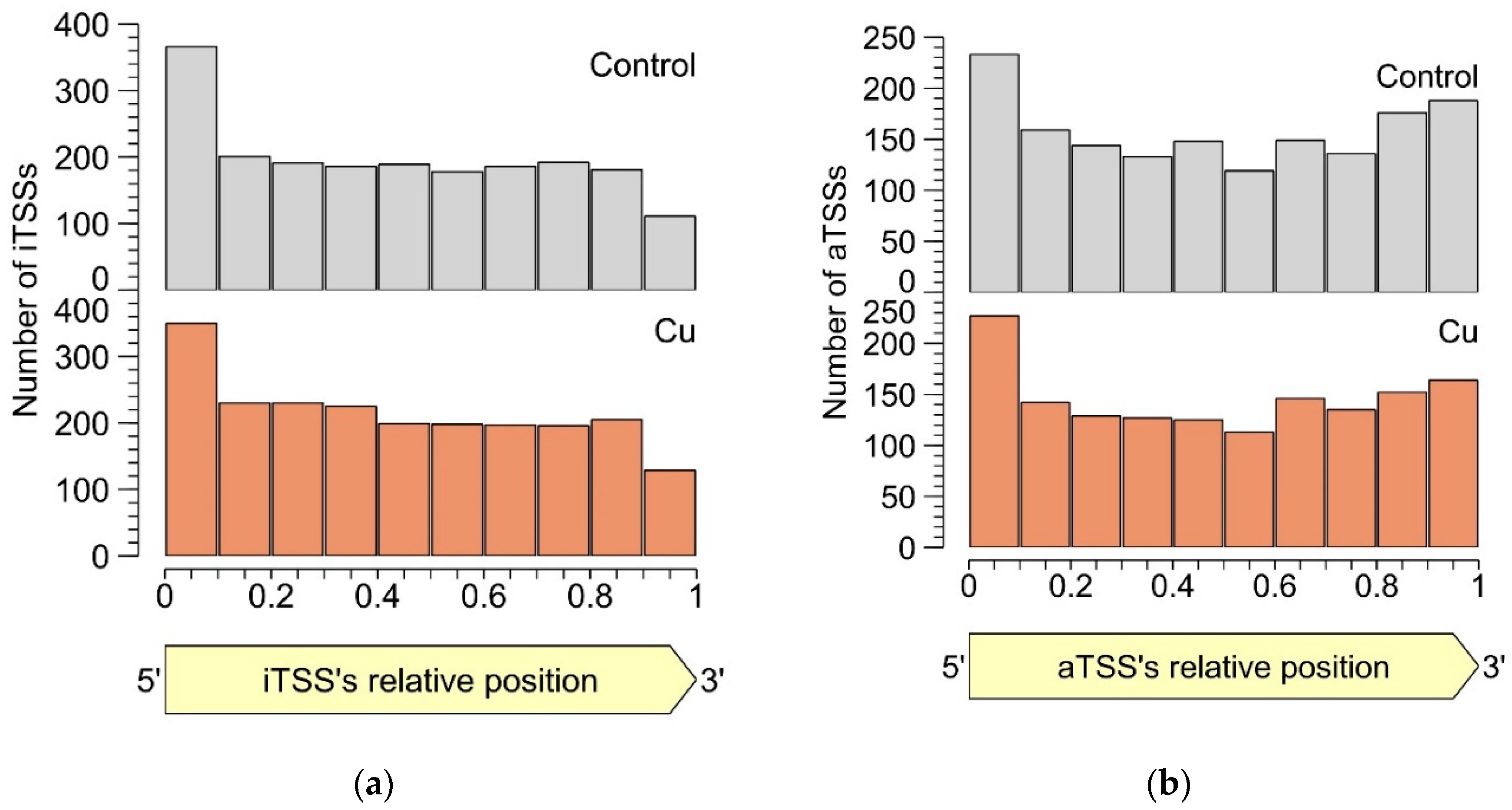
3.2.4. Intragenic TSSs
3.2.5. Antisense TSSs
3.2.6. Orphan TSSs
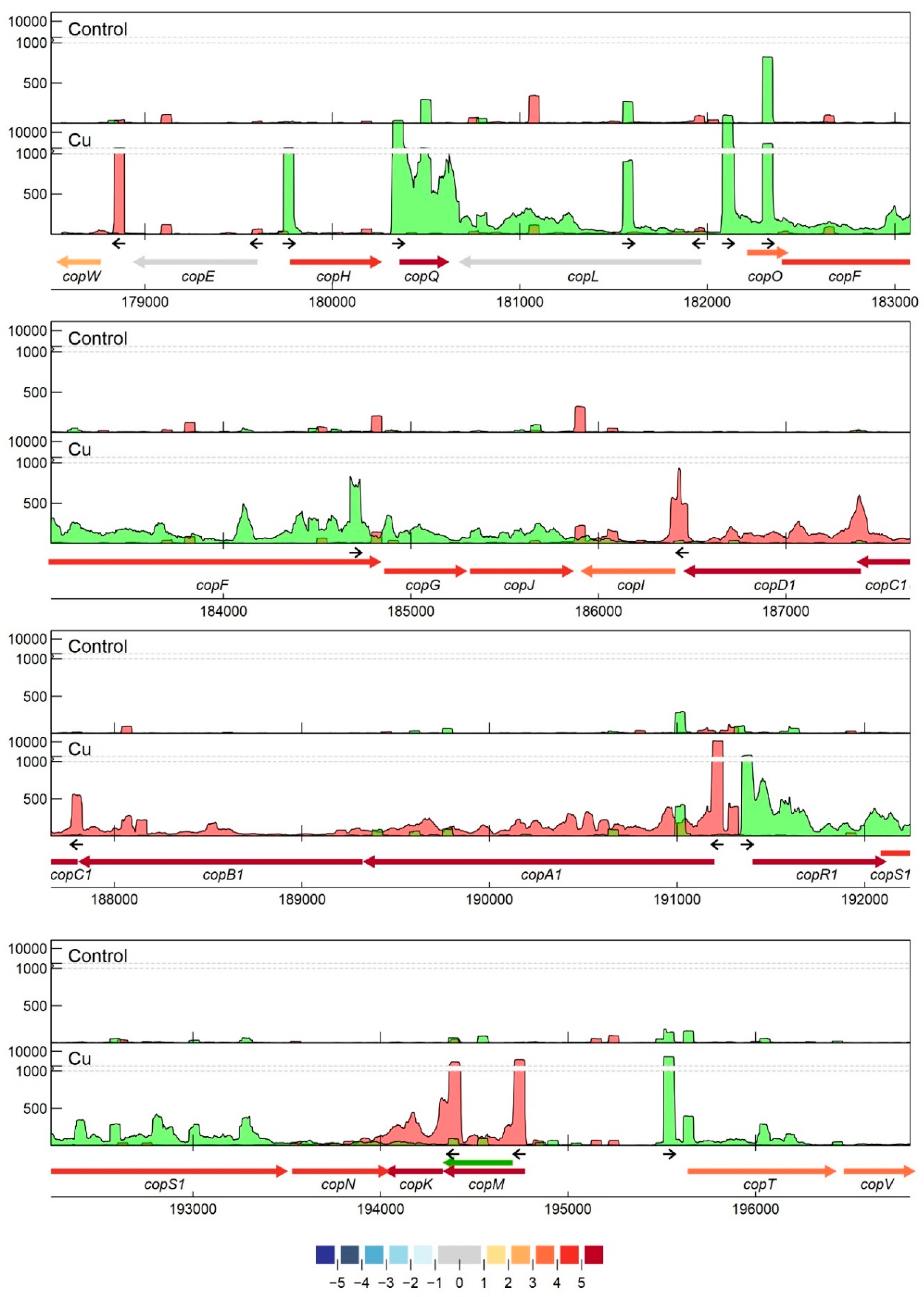
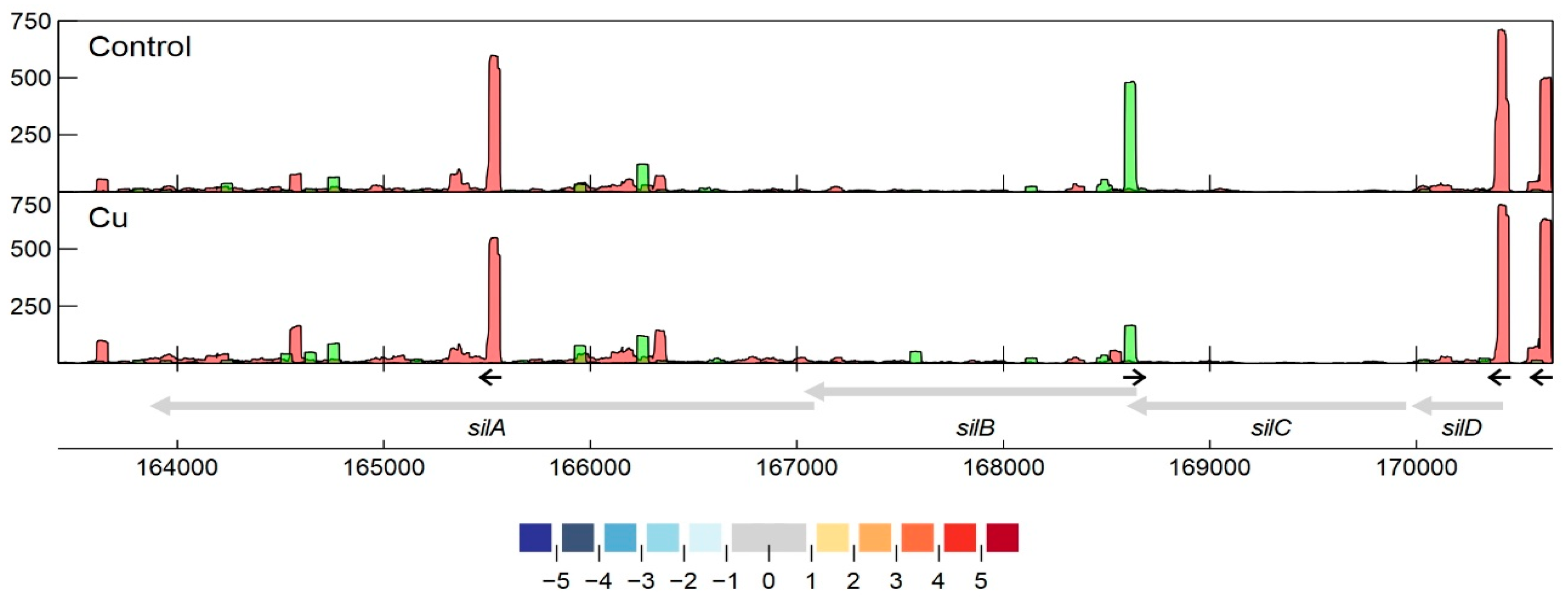
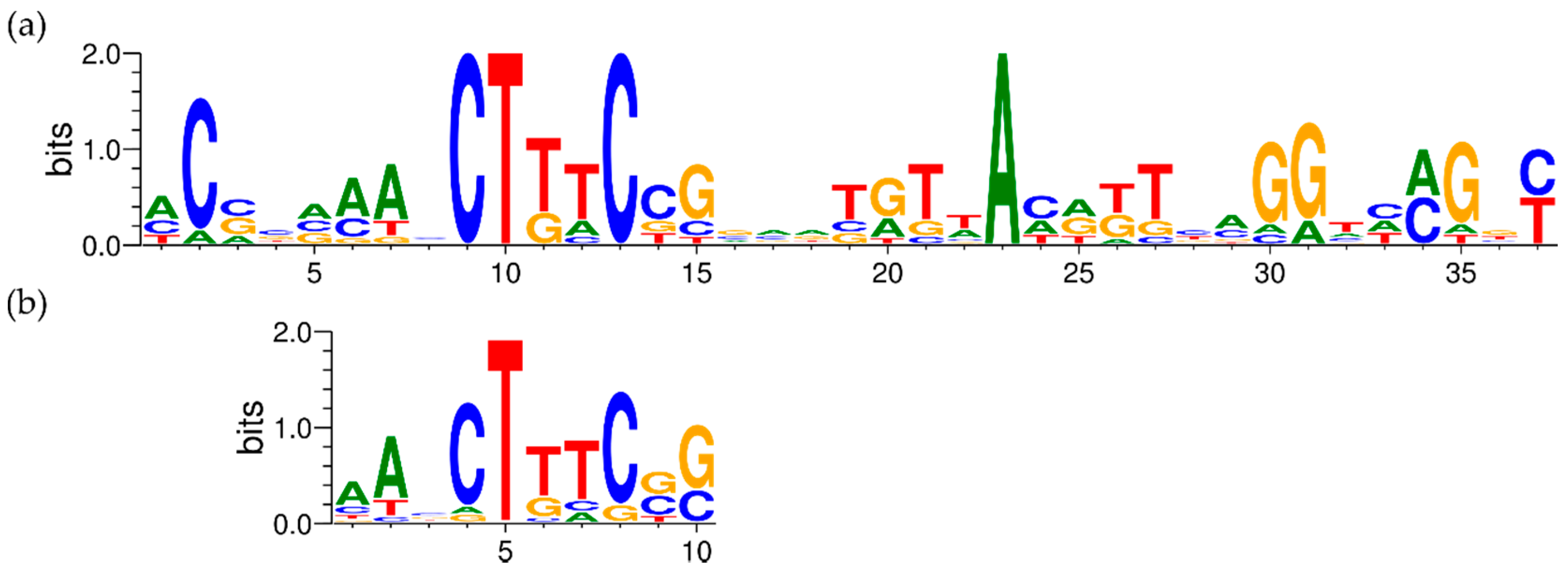
3.2.7. Detailed Analysis of cop and sil Clusters on pMOL30
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Valette-Silver, N.J. The Use of Sediment Cores to Reconstruct Historical Trends in Contamination of Estuarine and Coastal Sediments. Estuaries 1993, 16, 577–588. [Google Scholar] [CrossRef]
- Nicholson, F.A.; Smith, S.; Alloway, B.; Carlton-Smith, C.; Chambers, B. An inventory of heavy metals inputs to agricultural soils in England and Wales. Sci. Total. Environ. 2003, 311, 205–219. [Google Scholar] [CrossRef]
- François, F.; Lombard, C.; Guigner, J.-M.; Soreau, P.; Brian-Jaisson, F.; Martino, G.; Vandervennet, M.; Garcia, D.; Molinier, A.-L.; Pignol, D.; et al. Isolation and Characterization of Environmental Bacteria Capable of Extracellular Biosorption of Mercury. Appl. Environ. Microbiol. 2011, 78, 1097–1106. [Google Scholar] [CrossRef] [PubMed]
- Samanta, A.; Bera, P.; Khatun, M.; Sinha, C.; Pal, P.; Lalee, A.; Mandal, A. An investigation on heavy metal tolerance and antibiotic resistance properties of bacterial strain Bacillus sp. isolated from municipal waste. J. Microbiol. Biotech. Res. 2012, 2, 178–189. [Google Scholar]
- Sobolev, D.; Begonia, M.F.T. Effects of Heavy Metal Contamination upon Soil Microbes: Lead-induced Changes in General and Denitrifying Microbial Communities as Evidenced by Molecular Markers. Int. J. Environ. Res. Public Heal. 2008, 5, 450–456. [Google Scholar] [CrossRef]
- Hoostal, M.J.; Bidart-Bouzat, M.G.; Bouzat, J.L. Local adaptation of microbial communities to heavy metal stress in polluted sediments of Lake Erie. FEMS Microbiol. Ecol. 2008, 65, 156–168. [Google Scholar] [CrossRef]
- Berg, J.; Brandt, K.K.; Abu Al-Soud, W.; Holm, P.E.; Hansen, L.H.; Sørensen, S.J.; Nybroe, O. Selection for Cu-Tolerant Bacterial Communities with Altered Composition, but Unaltered Richness, via Long-Term Cu Exposure. Appl. Environ. Microbiol. 2012, 78, 7438–7446. [Google Scholar] [CrossRef]
- Igiri, B.E.; Okoduwa, S.I.R.; Idoko, G.O.; Akabuogu, E.P.; Adeyi, A.O.; Ejiogu, I.K. Toxicity and Bioremediation of Heavy Metals Contaminated Ecosystem from Tannery Wastewater: A Review. J. Toxicol. 2018, 2018, 1–16. [Google Scholar] [CrossRef]
- Dickinson, A.; Power, A.; Hansen, M.; Brandt, K.; Piliposian, G.; Appleby, P.; O’Neill, P.; Jones, R.; Sierocinski, P.; Koskella, B.; et al. Heavy metal pollution and co-selection for antibiotic resistance: A microbial palaeontology approach. Environ. Int. 2019, 132, 105117. [Google Scholar] [CrossRef]
- Köhler, S.; Belkin, S.; Schmid, R.D. Reporter gene bioassays in environmental analysis. Anal. Bioanal. Chem. 2001, 366, 769–779. [Google Scholar] [CrossRef]
- Tauriainen, S.; Virta, M.; Karp, M. Detecting bioavailable toxic metals and metalloids from natural water samples using luminescent sensor bacteria. Water Res. 2000, 34, 2661–2666. [Google Scholar] [CrossRef]
- Flynn, H.; Meharg, A.; Bowyer, P.K.; Paton, G. Antimony bioavailability in mine soils. Environ. Pollut. 2003, 124, 93–100. [Google Scholar] [CrossRef]
- Magrisso, S.; Erel, Y.; Belkin, S. Microbial reporters of metal bioavailability. Microb. Biotechnol. 2008, 1, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Rensing, C.; McDevitt, S.F. The copper metallome in prokaryotic cells. Met. Ions Life Sci. 2013, 12, 417–450. [Google Scholar]
- Meliani, A.; Bensoltane, A. Biofilm-Mediated Heavy Metals Bioremediation in PGPR Pseudomonas. J. Bioremediation Biodegrad. 2016, 7. [Google Scholar] [CrossRef]
- Xu, Z.; Lei, Y.; Patel, J. Bioremediation of soluble heavy metals with recombinant Caulobacter crescentus. Bioeng. Bugs 2010, 1, 207–212. [Google Scholar] [CrossRef]
- Tabak, H.H.; Lens, P.N.L.; Van Hullebusch, E.D.; Dejonghe, W. Developments in Bioremediation of Soils and Sediments Polluted with Metals and Radionuclides–1. Microbial Processes and Mechanisms Affecting Bioremediation of Metal Contamination and Influencing Metal Toxicity and Transport. Rev. Environ. Sci. Biotechnol. 2005, 4, 115–156. [Google Scholar] [CrossRef]
- Akcil, A.; Erust, C.; Ozdemiroglu, S.; Fonti, V.; Beolchini, F. A review of approaches and techniques used in aquatic contaminated sediments: Metal removal and stabilization by chemical and biotechnological processes. J. Clean. Prod. 2015, 86, 24–36. [Google Scholar] [CrossRef]
- Mergeay, M.; Van Houdt, R. Metal Response in Cupriavidus metallidurans Volume I: From Habitats to Genes and Proteins; Springer: Berlin, Germany, 2015. [Google Scholar]
- Mergeay, M.; Houba, C.; Gerits, J. Extrachromosomal Inheritance Controlling Resistance to Cobalt, Cadmium, Copper, and Zinc Ions: Evidence from Curing in a Pseudomonas. Arch. Int. Physiol. Biochim. 1978, 86, 440–441. [Google Scholar]
- Mergeay, M.; Nies, D.; Schlegel, H.G.; Gerits, J.; Charles, P.; Van Gijsegem, F. Alcaligenes eutrophus CH34 is a facultative chemolithotroph with plasmid-bound resistance to heavy metals. J. Bacteriol. 1985, 162, 328–334. [Google Scholar] [CrossRef]
- Monsieurs, P.; Moors, H.; Van Houdt, R.; Janssen, P.J.; Janssen, A.; Coninx, I.; Mergeay, M.; Leys, N. Heavy metal resistance in Cupriavidus metallidurans CH34 is governed by an intricate transcriptional network. BioMetals 2011, 24, 1133–1151. [Google Scholar] [CrossRef] [PubMed]
- Nies, D.H. Microbial heavy-metal resistance. Appl. Microbiol. Biotechnol. 1999, 51, 730–750. [Google Scholar] [CrossRef] [PubMed]
- Diels, L.; Van Roy, S.; Taghavi, S.; Van Houdt, R. From industrial sites to environmental applications with Cupriavidus metallidurans. Antonie Van Leeuwenhoek 2009, 96, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Collard, J.M.; Corbisier, P.; Diels, L.; Dong, Q.; Jeanthon, C.; Mergeay, M.; Taghavi, S.; van der Lelie, D.; Wilmotte, A.; Wuertz, S. Plasmids for heavy metal resistance in Alcaligenes eutrophus CH34: Mechanisms and applications. FEMS Microbiol. Rev. 1994, 14, 405–414. [Google Scholar] [CrossRef]
- Corbisier, P.; Thiry, E.; Diels, L. Bacterial Biosensors for the Toxicity Assessment of Solid Wastes. Environ Tox. Water Qual. Int. J. 1996, 11, 171–177. [Google Scholar] [CrossRef]
- Tibazarwa, C.; Corbisier, P.; Mench, M.; Bossus, A.; Solda, P.; Mergeay, M.; Wyns, L.; Van Der Lelie, D. A microbial biosensor to predict bioavailable nickel in soil and its transfer to plants. Environ. Pollut. 2001, 113, 19–26. [Google Scholar] [CrossRef]
- Corbisier, P.; Van Der Lelie, D.; Borremans, B.; Provoost, A.; De Lorenzo, V.; Brown, N.L.; Lloyd, J.R.; Hobman, J.L.; Csöregi, E.; Johansson, G.; et al. Whole cell- and protein-based biosensors for the detection of bioavailable heavy metals in environmental samples. Anal. Chim. Acta 1999, 387, 235–244. [Google Scholar] [CrossRef]
- Diels, L.; De Smet, M.; Hooyberghs, L.; Corbisier, P. Heavy Metals Bioremediation of Soil. Mol. Biotechnol. 1999, 12, 149–158. [Google Scholar] [CrossRef]
- Berezina, N.; Yada, B.; Lefebvre, R. From organic pollutants to bioplastics: Insights into the bioremediation of aromatic compounds by Cupriavidus necator. N. Biotechnol. 2015, 32, 47–53. [Google Scholar] [CrossRef]
- Janssen, P.J.; Van Houdt, R.; Moors, H.; Monsieurs, P.; Morin, N.; Michaux, A.; Benotmane, M.A.; Leys, N.; Vallaeys, T.; Lapidus, A.; et al. The Complete Genome Sequence of Cupriavidus metallidurans Strain CH34, a Master Survivalist in Harsh and Anthropogenic Environments. PLoS ONE 2010, 5, e10433. [Google Scholar] [CrossRef]
- Lee, K.-Y.; Bosch, J.; Meckenstock, R.U. Use of metal-reducing bacteria for bioremediation of soil contaminated with mixed organic and inorganic pollutants. Environ. Geochem. Heal. 2011, 34, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Köhler, C.; Lourenco, R.; Avelar, G.M.; Gomes, S.L. Extracytoplasmic function (ECF) sigma factor σF is involved in Caulobacter crescentus response to heavy metal stress. BMC Microbiol. 2012, 12, 210. [Google Scholar] [CrossRef] [PubMed]
- Große, C.; Poehlein, A.; Blank, K.; Schwarzenberger, C.; Schleuder, G.; Herzberg, M.; Nies, D.H.; Grosse, C. The third pillar of metal homeostasis in Cupriavidus metallidurans CH34: Preferences are controlled by extracytoplasmic function sigma factors. Metallomics 2019, 11, 291–316. [Google Scholar] [CrossRef]
- Osman, D.; Cavet, J.S. Copper Homeostasis in Bacteria. Adv. Appl. Microbiol. 2008, 65, 217–247. [Google Scholar] [PubMed]
- Argüello, J.M.; Raimunda, D.; Padilla-Benavides, T. Mechanisms of copper homeostasis in bacteria. Front. Microbiol. 2013, 3, 73. [Google Scholar] [CrossRef]
- Holmqvist, E.; Wagner, E.G.H. Impact of bacterial sRNAs in stress responses. Biochem. Soc. Trans. 2017, 45, 1203–1212. [Google Scholar] [CrossRef]
- Hör, J.; Vogel, J. Global snapshots of bacterial RNA networks. EMBO J. 2016, 36, 245–247. [Google Scholar] [CrossRef][Green Version]
- Chen, J.; Morita, T.; Gottesman, S. Regulation of Transcription Termination of Small RNAs and by Small RNAs: Molecular Mechanisms and Biological Functions. Front. Microbiol. 2019, 9, 201. [Google Scholar] [CrossRef]
- Dietrich, M.; Munke, R.; Gottschald, M.; Ziska, E.; Boettcher, J.P.; Mollenkopf, H.-J.; Friedrich, A. The effect of hfq on global gene expression and virulence in Neisseria gonorrhoeae. FEBS J. 2009, 276, 5507–5520. [Google Scholar] [CrossRef]
- Sousa, S.A.; Ramos, C.G.; Moreira, L.M.; Leitão, J.H. The hfq gene is required for stress resistance and full virulence of Burkholderia cepacia to the nematode Caenorhabditis elegans. Microbiology 2009, 156, 896–908. [Google Scholar] [CrossRef]
- Feliciano, J.R.; Grilo, A.M.; I Guerreiro, S.; Sousa, S.A.; Leitão, J.H. Hfq: A multifaceted RNA chaperone involved in virulence. Futur. Microbiol. 2016, 11, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Kakoschke, T.K.; Kakoschke, S.C.; Zeuzem, C.; Bouabe, H.; Adler, K.; Heesemann, J.; Rossier, O. The RNA Chaperone Hfq Is Essential for Virulence and Modulates the Expression of Four Adhesins in Yersinia enterocolitica. Sci. Rep. 2016, 6, 29275. [Google Scholar] [CrossRef] [PubMed]
- Fantappiè, L.; Metruccio, M.; Seib, K.L.; Oriente, F.; Cartocci, E.; Ferlicca, F.; Giuliani, M.M.; Scarlato, V.; Delany, I. The RNA Chaperone Hfq Is Involved in Stress Response and Virulence in Neisseria meningitidis and Is a Pleiotropic Regulator of Protein Expression. Infect. Immun. 2009, 77, 1842–1853. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.W.; Ott, C.M.; Zu Bentrup, K.H.; Ramamurthy, R.; Quick, L.; Porwollik, S.; Cheng, P.; McClelland, M.; Tsaprailis, G.; Radabaugh, T.; et al. Space flight alters bacterial gene expression and virulence and reveals a role for global regulator Hfq. Proc. Natl. Acad. Sci. USA 2007, 104, 16299–16304. [Google Scholar] [CrossRef]
- Eshghi, A.; Becam, J.; Lambert, A.; Sismeiro, O.; Dillies, M.-A.; Jagla, B.; Wunder, E.A.J.; Ko, A.I.; Coppee, J.-Y.; Goarant, C.; et al. A Putative Regulatory Genetic Locus Modulates Virulence in the Pathogen Leptospira interrogans. Infect. Immun. 2014, 82, 2542–2552. [Google Scholar] [CrossRef]
- Geng, J.; Song, Y.; Yang, L.; Feng, Y.; Qiu, Y.; Li, G.; Guo, J.; Bi, Y.; Qu, Y.; Wang, W.; et al. Involvement of the Post-Transcriptional Regulator Hfq in Yersinia pestis Virulence. PLoS ONE 2009, 4, e6213. [Google Scholar] [CrossRef]
- Molina-Santiago, C.; Daddaoua, A.; Gómez-Lozano, M.; Udaondo, Z.; Molin, S.; Ramos, J.L. Differential transcriptional response to antibiotics by Pseudomonas putida DOT-T1E. Environ. Microbiol. 2015, 17, 3251–3262. [Google Scholar] [CrossRef]
- Chen, Y.; Indurthi, D.C.; Jones, S.W.; Papoutsakis, E.T. Small RNAs in the Genus Clostridium. mBio 2011, 2, 00340-10. [Google Scholar] [CrossRef]
- Yu, J.; Schneiders, T. Tigecycline challenge triggers sRNA production in Salmonella enterica serovar Typhimurium. BMC Microbiol. 2012, 12, 195. [Google Scholar] [CrossRef]
- Howden, B.P.; Beaume, M.; Harrison, P.F.; Hernandez, D.; Schrenzel, J.; Seemann, T.; Francois, P.; Stinear, T.P. Analysis of the Small RNA Transcriptional Response in Multidrug-Resistant Staphylococcus aureus after Antimicrobial Exposure. Antimicrob. Agents Chemother. 2013, 57, 3864–3874. [Google Scholar] [CrossRef]
- Kim, K.-S.; Bak, G.; Lee, J. Systematic analysis of the role of bacterial Hfq-interacting sRNAs in the response to antibiotics. J. Antimicrob. Chemother. 2015, 70, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, K.; Papenfort, K. Interplay of regulatory RNAs and mobile genetic elements in enteric pathogens. Mol. Microbiol. 2016, 101, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Oglesby-Sherrouse, A.G.; Murphy, E.R. Iron-responsive bacterial small RNAs: Variations on a theme. Metallomics 2013, 5, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Chareyre, S.; Mandin, P. Bacterial Iron Homeostasis Regulation by sRNAs. Regul. RNA Bact. Archaea 2018, 6, 267–281. [Google Scholar] [CrossRef]
- Sridhar, J.; Gayathri, M. Transcriptome based Identification of silver stress responsive sRNAs from Bacillus cereus ATCC14579. Bioinformation 2019, 15, 474–479. [Google Scholar] [CrossRef]
- De Freitas, E.C.; Ucci, A.P.; Teixeira, E.C.; Pedroso, G.A.; Hilario, E.; Zocca, V.F.B.; De Paiva, G.B.; Ferreira, H.; Pedrolli, D.B.; Bertolini, M.C. The copper-inducible copAB operon in Xanthomonas citri subsp. citri is regulated at transcriptional and translational levels. Microbiology 2019, 165, 355–365. [Google Scholar] [CrossRef]
- Lalaouna, D.; Baude, J.; Wu, Z.; Tomasini, A.; Chicher, J.; Marzi, S.; Vandenesch, F.; Romby, P.; Caldelari, I.; Moreau, K. RsaC sRNA modulates the oxidative stress response of Staphylococcus aureus during manganese starvation. Nucleic Acids Res. 2019, 47, 9871–9887. [Google Scholar] [CrossRef]
- Chen, Y.; Xue, D.; Sun, W.; Han, J.; Li, J.; Gao, R.; Zhou, Z.; Zhang, W.; Chen, M.; Lin, M.; et al. sRNA OsiA Stabilizes Catalase mRNA during Oxidative Stress Response of Deincoccus radiodurans R1. Microorganisms 2019, 7, 422. [Google Scholar] [CrossRef]
- Peng, T.; Kan, J.; Lun, J.; Hu, Z. Identification of novel sRNAs involved in oxidative stress response in the fish pathogen Vibrio alginolyticus by transcriptome analysis. J. Fish Dis. 2018, 42, 277–291. [Google Scholar] [CrossRef]
- Olaya-Abril, A.; Luque-Almagro, V.M.; Pérez, M.D.; López, C.M.; Amil-Ruiz, F.; Cabello, P.; Sáez, L.P.; Moreno-Vivián, C.; Roldán, M.D. Putative small RNAs controlling detoxification of industrial cyanide-containing wastewaters by Pseudomonas pseudoalcaligenes CECT5344. PLoS ONE 2019, 14, e0212032. [Google Scholar] [CrossRef]
- Innocenti, N.; Golumbeanu, M.; D’Herouel, A.F.; Lacoux, C.; Bonnin, R.A.; Kennedy, S.P.; Wessner, F.; Serror, P.; Bouloc, P.; Repoila, F.; et al. Whole-genome mapping of 5′ RNA ends in bacteria by tagged sequencing: A comprehensive view in Enterococcus faecalis. RNA 2015, 21, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.M.; Vogel, J. Differential RNA-seq: The approach behind and the biological insight gained. Curr. Opin. Microbiol. 2014, 19, 97–105. [Google Scholar] [CrossRef]
- Maes, A.; Gracia, C.; Innocenti, N.; Zhang, K.; Aurell, E.; Hajnsdorf, E. Landscape of RNA polyadenylation in E. coli. Nucleic Acids Res. 2017, 45, 2746–2756. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, N.; Repoila, F.; Aurell, E. Detection and quantitative estimation of spurious double stranded DNA formation during reverse transcription in bacteria using tagRNA-seq. RNA Biol. 2015, 12, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Yue, L.; Feng, D.; Qi, F.; Li, J.; Dong, X. Genome-wide mRNA processing in methanogenic archaea reveals post-transcriptional regulation of ribosomal protein synthesis. Nucleic Acids Res. 2017, 45, 7285–7298. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.R.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Jensen, L.J.; Julien, P.; Kuhn, M.; Von Mering, C.; Muller, J.; Doerks, T.; Bork, P. eggNOG: Automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 2007, 36, D250–D254. [Google Scholar] [CrossRef]
- Vallenet, D.; Belda, E.; Calteau, A.; Cruveiller, S.; Engelen, S.; Lajus, A.; Le Fèvre, F.; Longin, C.; Mornico, D.; Roche, D.; et al. MicroScope—An integrated microbial resource for the curation and comparative analysis of genomic and metabolic data. Nucleic Acids Res. 2012, 41, D636–D647. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Monchy, S.; Benotmane, M.A.; Wattiez, R.; Van Aelst, S.; Auquier, V.; Borremans, B.; Mergeay, M.; Taghavi, S.; Van Der Lelie, D.; Vallaeys, T. Transcriptomic and proteomic analyses of the pMOL30-encoded copper resistance in Cupriavidus metallidurans strain CH34. Microbiology 2006, 152, 1765–1776. [Google Scholar] [CrossRef]
- Zhang, Y.-B.; Monchy, S.; Greenberg, B.; Mergeay, M.; Gang, O.; Taghavi, S.; Van Der Lelie, D. ArsR arsenic-resistance regulatory protein from Cupriavidus metallidurans CH34. Antonie Van Leeuwenhoek 2009, 96, 161–170. [Google Scholar] [CrossRef]
- Dieppois, G.; Ducret, V.; Caille, O.; Perron, K. The Transcriptional Regulator CzcR Modulates Antibiotic Resistance and Quorum Sensing in Pseudomonas aeruginosa. PLoS ONE 2012, 7, e38148. [Google Scholar] [CrossRef]
- Urano, H.; Yoshida, M.; Ogawa, A.; Yamamoto, K.; Ishihama, A.; Ogasawara, H. Cross-regulation between two common ancestral response regulators, HprR and CusR, in Escherichia coli. Microbiology 2017, 163, 243–252. [Google Scholar] [CrossRef]
- Mijnendonckx, K.; Ali, M.; Provoost, A.; Janssen, P.J.; Mergeay, M.; Leys, N.; Charlier, D.; Monsieurs, P.; Van Houdt, R. Spontaneous mutation in the AgrRS two-component regulatory system of Cupriavidus metallidurans results in enhanced silver resistance. Metallomics 2019, 11, 1912–1924. [Google Scholar] [CrossRef] [PubMed]
- Caille, O.; Rossier, C.; Perron, K. A Copper-Activated Two-Component System Interacts with Zinc and Imipenem Resistance in Pseudomonas aeruginosa. J. Bacteriol. 2007, 189, 4561–4568. [Google Scholar] [CrossRef] [PubMed]
- Reith, F.; Etschmann, B.; Grosse, C.; Moors, H.; Benotmane, M.A.; Monsieurs, P.; Grass, G.; Doonan, C.; Vogt, S.; Lai, B.; et al. Mechanisms of gold biomineralization in the bacterium Cupriavidus metallidurans. Proc. Natl. Acad. Sci. USA 2009, 106, 17757–17762. [Google Scholar] [CrossRef]
- Lemire, J.A.; Harrison, J.J.; Turner, R.J. Antimicrobial activity of metals: Mechanisms, molecular targets and applications. Nat. Rev. Genet. 2013, 11, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Mao, J.; Zhao, Y.; Hu, M.; Wang, X. Multiple Transcriptional Mechanisms Collectively Mediate Copper Resistance in Cupriavidus gilardii CR3. Environ. Sci. Technol. 2019, 53, 4609–4618. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.O.; Merchant, K.; Nudelman, R.; Beyer, W.F., Jr.; Keng, T.; De Angelo, J.; Hausladen, A.; Stamler, J.S. OxyR: A Molecular Code for Redox-Related Signaling. Cell 2002, 109, 383–396. [Google Scholar] [CrossRef]
- Koo, M.-S.; Lee, J.; Rah, S.; Yeo, W.; Lee, J.-W.; Lee, K.-L.; Koh, Y.; Kang, S.; Roe, J.-H. A reducing system of the superoxide sensor SoxR in Escherichia coli. EMBO J. 2003, 22, 2614–2622. [Google Scholar] [CrossRef]
- Lesniak, J.; Barton, W.A.; Nikolov, D.B. Structural and functional characterization of the Pseudomonas hydroperoxide resistance protein Ohr. EMBO J. 2002, 21, 6649–6659. [Google Scholar] [CrossRef]
- Sukchawalit, R.; Loprasert, S.; Atichartpongkul, S.; Mongkolsuk, S. Complex Regulation of the Organic Hydroperoxide Resistance Gene (ohr) from Xanthomonas Involves OhrR, a Novel Organic Peroxide-Inducible Negative Regulator, and Posttranscriptional Modifications. J. Bacteriol. 2001, 183, 4405–4412. [Google Scholar] [CrossRef]
- Scarpa, M.; Momo, F.; Viglino, P.; Vianello, F.; Rigo, A. Activated oxygen species in the oxidation of glutathione A kinetic study. Biophys. Chem. 1996, 60, 53–61. [Google Scholar] [CrossRef]
- Romsang, A.; Duang-Nkern, J.; Leesukon, P.; Saninjuk, K.; Vattanaviboon, P.; Mongkolsuk, S. The Iron-Sulphur Cluster Biosynthesis Regulator IscR Contributes to Iron Homeostasis and Resistance to Oxidants in Pseudomonas aeruginosa. PLoS ONE 2014, 9, e86763. [Google Scholar] [CrossRef] [PubMed]
- Chillappagari, S.; Seubert, A.; Trip, H.; Kuipers, O.P.; Marahiel, M.A.; Miethke, M. Copper Stress Affects Iron Homeostasis by Destabilizing Iron-Sulfur Cluster Formation in Bacillus subtilis. J. Bacteriol. 2010, 192, 2512–2524. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Dos Santos, P. Protective role of bacillithiol in superoxide stress and Fe-S metabolism in Bacillus subtilis. Microbiology 2015, 4, 616–631. [Google Scholar] [CrossRef]
- Fung, D.K.C.; Lau, W.Y.; Chan, W.T.; Yan, A. Copper Efflux Is Induced during Anaerobic Amino Acid Limitation in Escherichia coli To Protect Iron-Sulfur Cluster Enzymes and Biogenesis. J. Bacteriol. 2013, 195, 4556–4568. [Google Scholar] [CrossRef] [PubMed]
- Sticht, H.; Rösch, P. The structure of iron–sulfur proteins. Prog. Biophys. Mol. Biol. 1998, 70, 95–136. [Google Scholar] [CrossRef]
- Beinert, H. Iron-sulfur proteins: Ancient structures, still full of surprises. JBIC J. Biol. Inorg. Chem. 2000, 5, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Woo, K.M.; I Kim, K.; Goldberg, A.L.; Ha, D.B.; Chung, C.H. The heat-shock protein ClpB in Escherichia coli is a protein-activated ATPase. J. Biol. Chem. 1992, 267, 20429–20434. [Google Scholar]
- Skowyra, D.; Georgopoulos, C.; Zylicz, M. The E. coli dnaK Gene Product, the hsp70 Homolog, Can Reactivate Heat-Inactivated RNA Polymerase in an ATP Hydrolysis-independent Manner. Cell 1990, 62, 939–944. [Google Scholar] [CrossRef]
- Rohrwild, M.; Coux, O.; Huang, H.C.; Moerschell, R.P.; Yoo, S.J.; Seol, J.H.; Chung, C.H.; Goldberg, A.L. HslV-HslU: A novel ATP-dependent protease complex in Escherichia coli related to the eukaryotic proteasome. Proc. Natl. Acad. Sci. USA 1996, 93, 5808–5813. [Google Scholar] [CrossRef]
- Gutsche, J.; Remminghorst, U.; Rehm, B.H.A. Biochemical analysis of alginate biosynthesis protein AlgX from Pseudomonas aeruginosa: Purification of an AlgX-MucD (AlgY) protein complex. Biochimie 2006, 88, 245–251. [Google Scholar] [CrossRef]
- Irisawa, T.; Kato, M.; Moroishi, J.; Muramatu, M. Effect of trans-4-Guanidinomethylcyclohexanecarboxylic Acid 4-tert-Butylphenyl Ester, a Trypsin Inhibitor, on the Growth of Various Strains of Escherichia coli. Biol. Pharm. Bull. 1993, 16, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Bieniossek, C.; Schalch, T.; Bumann, M.; Meister, M.; Meier, R.; Baumann, U. The molecular architecture of the metalloprotease FtsH. Proc. Natl. Acad. Sci. USA 2006, 103, 3066–3071. [Google Scholar] [CrossRef] [PubMed]
- Tomoyasu, T.; Gamer, J.; Bukau, B.; Kanemori, M.; Mori, H.; Rutman, A.; Oppenheim, A.B.; Yura, T.; Yamanaka, K.; Niki, H. Escherichia coli FtsH is a membrane-bound, ATP-dependent protease which degrades the heat-shock transcription factor sigma 32. EMBO J. 1995, 14, 2551–2560. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.H.; Solioz, M. Copper-induced Proteolysis of the CopZ Copper Chaperone of Enterococcus hirae. J. Biol. Chem. 2001, 276, 47822–47827. [Google Scholar] [CrossRef]
- Solioz, M. Role of proteolysis in copper homoeostasis. Biochem. Soc. Trans. 2002, 30, 688–691. [Google Scholar] [CrossRef]
- Van Der Ploeg, J.R.; Eichhorn, E.; Leisinger, T. Sulfonate-sulfur metabolism and its regulation in Escherichia coli. Arch. Microbiol. 2001, 176, 1–8. [Google Scholar] [CrossRef]
- Salazar, C.N.; Acosta, M.; Galleguillos, P.A.; Shmaryahu, A.; Quatrini, R.; Holmes, D.S.; Demergasso, C. Analysis of Gene Expression in Response to Copper Stress in Acidithiobacillus ferrooxidans Strain D2, Isolated from a Copper Bioleaching Operation. Adv. Mater. Res. 2013, 825, 157–161. [Google Scholar] [CrossRef]
- Wheaton, G.H.; Mukherjee, A.; Kelly, R.M. Transcriptomes of the Extremely Thermoacidophilic Archaeon Metallosphaera sedula Exposed to Metal “Shock” Reveal Generic and Specific Metal Responses. Appl. Environ. Microbiol. 2016, 82, 4613–4627. [Google Scholar] [CrossRef]
- Huang, N.; Mao, J.; Hu, M.; Wang, X.; Huo, M. Responses to copper stress in the metal-resistant bacterium Cupriavidus gilardii CR3: A whole-transcriptome analysis. J. Basic Microbiol. 2019, 59, 446–457. [Google Scholar] [CrossRef]
- Rigo, A.; Corazza, A.; Di Paolo, M.L.; Rossetto, M.; Ugolini, R.; Scarpa, M. Interaction of copper with cysteine: Stability of cuprous complexes and catalytic role of cupric ions in anaerobic thiol oxidation. J. Inorg. Biochem. 2004, 98, 1495–1501. [Google Scholar] [CrossRef]
- Heidrich, N.; Bauriedl, S.; Barquist, L.; Li, L.; Schoen, C.; Vogel, J. The primary transcriptome of Neisseria meningitidis and its interaction with the RNA chaperone Hfq. Nucleic Acids Res. 2017, 45, 6147–6167. [Google Scholar] [CrossRef] [PubMed]
- Vandecraen, J.; Monsieurs, P.; Mergeay, M.; Leys, N.; Aertsen, A.; Van Houdt, R. Zinc-Induced Transposition of Insertion Sequence Elements Contributes to Increased Adaptability of Cupriavidus metallidurans. Front. Microbiol. 2016, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Borremans, B.; Hobman, J.L.; Provoost, A.; Brown, N.L.; Van Der Lelie, D. Cloning and Functional Analysis of thepbr Lead Resistance Determinant of Ralstonia metallidurans CH34. J. Bacteriol. 2001, 183, 5651–5658. [Google Scholar] [CrossRef] [PubMed]
- Große, C.; Grass, G.; Anton, A.; Franke, S.; Santos, A.N.; Lawley, B.; Brown, N.L.; Nies, D.H. Transcriptional Organization of the czcHeavy-Metal Homeostasis Determinant from Alcaligenes eutrophus. J. Bacteriol. 1999, 181, 2385–2393. [Google Scholar] [CrossRef]
- Lomsadze, A.; Gemayel, K.; Tang, S.; Borodovsky, M. Modeling leaderless transcription and atypical genes results in more accurate gene prediction in prokaryotes. Genome Res. 2018, 28, 1079–1089. [Google Scholar] [CrossRef]
- Schmidt, T.; Schlegel, H.G. Combined nickel-cobalt-cadmium resistance encoded by the ncc locus of Alcaligenes xylosoxidans 31A. J. Bacteriol. 1994, 176, 7045–7054. [Google Scholar] [CrossRef] [PubMed]
- Hynninen, A.; Touzé, T.; Pitkänen, L.; Mengin-Lecreulx, D.; Virta, M.; Mengin-Lecreulx, M. An efflux transporter PbrA and a phosphatase PbrB cooperate in a lead-resistance mechanism in bacteria. Mol. Microbiol. 2009, 74, 384–394. [Google Scholar] [CrossRef]
- Keshav, V.; Achilonu, I.; Dirr, H.W.; Kondiah, K. Recombinant expression and purification of a functional bacterial metallo-chaperone PbrD-fusion construct as a potential biosorbent for Pb(II). Protein Expr. Purif. 2019, 158, 27–35. [Google Scholar] [CrossRef]
- Sass, A.M.; Van Acker, H.; Förstner, K.U.; Van Nieuwerburgh, F.; Deforce, D.; Vogel, J.; Coenye, T. Genome-wide transcription start site profiling in biofilm-grown Burkholderia cenocepacia J2315. BMC Genom. 2015, 16, 775. [Google Scholar] [CrossRef]
- Vockenhuber, M.-P.; Sharma, C.M.; Statt, M.G.; Schmidt, D.; Xu, Z.; Dietrich, S.; Liesegang, H.; Mathews, D.H.; Suess, B. Deep sequencing-based identification of small non-coding RNAs in Streptomyces coelicolor. RNA Biol. 2011, 8, 468–477. [Google Scholar] [CrossRef][Green Version]
- Kröger, C.; Dillon, S.C.; Cameron, A.D.S.; Papenfort, K.; Sivasankaran, S.K.; Hokamp, K.; Chao, Y.; Sittka, A.; Hébrard, M.; Haendler, K.; et al. The transcriptional landscape and small RNAs of Salmonella enterica serovar Typhimurium. Proc. Natl. Acad. Sci. USA 2012, 109, E1277–E1286. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Schrader, J.M.; Kalogeraki, V.S.; Abeliuk, E.; Dinh, C.B.; Pham, J.Q.; Cui, Z.Z.; Dill, D.L.; McAdams, H.H.; Shapiro, L. The Global Regulatory Architecture of Transcription during the Caulobacter Cell Cycle. PLoS Genet. 2015, 11, e1004831. [Google Scholar] [CrossRef] [PubMed]
- Schrader, J.M.; Zhou, B.; Li, G.-W.; Lasker, K.; Childers, W.S.; Williams, B.; Long, T.; Crosson, S.; McAdams, H.H.; Weissman, J.S.; et al. The Coding and Noncoding Architecture of the Caulobacter crescentus Genome. PLoS Genet. 2014, 10, e1004463. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.M.; Hoffmann, S.; Darfeuille, F.; Reignier, J.; Findeiß, S.; Sittka, A.; Chabas, S.; Reiche, K.; Hackermüller, J.; Reinhardt, R.; et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 2010, 464, 250–255. [Google Scholar] [CrossRef]
- Bischler, T.; Tan, H.S.; Nieselt, K.; Sharma, C.M. Differential RNA-seq (dRNA-seq) for annotation of transcriptional start sites and small RNAs in Helicobacter pylori. Methods 2015, 86, 89–101. [Google Scholar] [CrossRef]
- Zheng, X.; Hu, G.; She, Z.-S.; Zhu, H. Leaderless genes in bacteria: Clue to the evolution of translation initiation mechanisms in prokaryotes. BMC Genom. 2011, 12, 361. [Google Scholar] [CrossRef]
- Beck, H.J.; Moll, I. Leaderless mRNAs in the Spotlight: Ancient but Not Outdated! Microbiol. Spectr. 2018, 6, 155–170. [Google Scholar] [CrossRef]
- Ao, W.; Gaudet, J.; Kent, W.J.; Muttumu, S.; Mango, S.E. Environmentally Induced Foregut Remodeling by PHA-4/FoxA and DAF-12/NHR. Science 2004, 305, 1743–1746. [Google Scholar] [CrossRef]
- Li, J.; Qi, L.; Guo, Y.; Yue, L.; Li, Y.; Ge, W.; Wu, J.; Shi, W.; Dong, X. Global mapping transcriptional start sites revealed both transcriptional and post-transcriptional regulation of cold adaptation in the methanogenic archaeon Methanolobus psychrophilus. Sci. Rep. 2015, 5, 9209. [Google Scholar] [CrossRef]
- Mitschke, J.; Vioque, A.; Haas, F.B.; Hess, W.R.; Muro-Pastor, A.M. Dynamics of transcriptional start site selection during nitrogen stress-induced cell differentiation in Anabaena sp. PCC7120. Proc. Natl. Acad. Sci. USA 2011, 108, 20130–20135. [Google Scholar] [CrossRef]
- Thomason, M.K.; Bischler, T.; Eisenbart, S.K.; Förstner, K.U.; Zhang, A.; Herbig, A.; Nieselt, K.; Sharma, C.M.; Storz, G. Global Transcriptional Start Site Mapping Using Differential RNA Sequencing Reveals Novel Antisense RNAs in Escherichia coli. J. Bacteriol. 2014, 197, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.-H.; Lee, S.-J.; Cho, B.; Lee, Y. Differential promoter usage ofinfAin response to cold shock in Escherichia coli. FEBS Lett. 2005, 580, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Güell, M.; Yus, E.; Lluch-Senar, M.; Serrano, L. Bacterial transcriptomics: What is beyond the RNA horiz-ome? Nat. Rev. Genet. 2011, 9, 658–669. [Google Scholar] [CrossRef]
- Lloréns-Rico, V.; Cano, J.; Kamminga, T.; Gil Garcia, R.; Latorre, A.; Chen, W.-H.; Bork, P.; Glass, J.I.; Serrano, L.; Lluch-Senar, M. Bacterial antisense RNAs are mainly the product of transcriptional noise. Sci. Adv. 2016, 2, e1501363. [Google Scholar] [CrossRef] [PubMed]
- Thomason, M.K.; Storz, G. Bacterial Antisense RNAs: How Many Are There, and What Are They Doing? Annu. Rev. Genet. 2010, 44, 167–188. [Google Scholar] [CrossRef]
- Eckweiler, D.; Häussler, S. Antisense transcription in Pseudomonas aeruginosa. Microbiology 2018, 164, 889–895. [Google Scholar] [CrossRef]
- Nicolas, P.; Mäder, U.; Dervyn, E.; Rochat, T.; LeDuc, A.; Pigeonneau, N.; Bidnenko, E.; Marchadier, E.; Hoebeke, M.; Aymerich, S.; et al. Condition-Dependent Transcriptome Reveals High-Level Regulatory Architecture in Bacillus subtilis. Science 2012, 335, 1103–1106. [Google Scholar] [CrossRef]
- Eiamphungporn, W.; Helmann, J.D. Extracytoplasmic function sigma factors regulate expression of the Bacillus subtilis yabE gene via a cis-acting antisense RNA. J. Bacteriol. 2009, 191, 1101–1105. [Google Scholar] [CrossRef]
- Madjalani, N.; Cunning, C.; Sledjeski, D.; Elliott, T.; Gottesman, S. DsrA RNA regulates translation of RpoS message by an anti-antisense mechanism, independent of its action as an antisilencer of transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 12462–12467. [Google Scholar]
- Friedman, R.C.; Kalkhof, S.; Doppelt-Azeroual, O.; Mueller, S.A.; Chovancová, M.; Von Bergen, M.; Schwikowski, B. Common and phylogenetically widespread coding for peptides by bacterial small RNAs. BMC Genom. 2017, 18, 553. [Google Scholar] [CrossRef]
- Tsai, C.-H.; Liao, R.; Chou, B.; Palumbo, M.; Contreras, L.M. Genome-Wide Analyses in Bacteria Show Small-RNA Enrichment for Long and Conserved Intergenic Regions. J. Bacteriol. 2014, 197, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Sobrero, P.; Valverde, C. The bacterial protein Hfq: Much more than a mere RNA-binding factor. Crit. Rev. Microbiol. 2012, 38, 276–299. [Google Scholar] [CrossRef] [PubMed]
- Staehlin, B.M.; Gibbons, J.G.; Rokas, A.; O’Halloran, T.V.; Slot, J.C. Evolution of a Heavy Metal Homeostasis/Resistance Island Reflects Increasing Copper Stress in Enterobacteria. Genome Biol. Evol. 2016, 8, 811–826. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar] [PubMed]
- Galperin, M.Y. Structural Classification of Bacterial Response Regulators: Diversity of Output Domains and Domain Combinations. J. Bacteriol. 2006, 188, 4169–4182. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Provoost, A.; Mijnendonckx, K.; Van Houdt, R.; Charlier, D. DNA-Binding and Transcription Activation by Unphosphorylated Response Regulator AgrR From Cupriavidus metallidurans Involved in Silver Resistance. Front. Microbiol. 2020, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef]
- Sass, A.M.; Kiekens, S.; Coenye, T. Identification of small RNAs abundant in Burkholderia cenocepacia biofilms reveal putative regulators with a potential role in carbon and iron metabolism. Sci. Rep. 2017, 7, 15665. [Google Scholar] [CrossRef]
- Hot, D.; Slupek, S.; Wulbrecht, B.; D’Hondt, A.; Hubans, C.; Antoine, R.; Locht, C.; Lemoine, Y. Detection of small RNAs in Bordetella pertussis and identification of a novel repeated genetic element. BMC Genom. 2011, 12, 1–13. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Cluster | Locus Tag 1 | Replicon 2 | Homologous Gene Cluster | Locus Tag 1 | Replicon 2 | Function |
|---|---|---|---|---|---|---|
| copA1B1C1D1 | 6112–6115 | pMOL30 | copA2B2C2D2 | 5671–5668 | CHR2 | Periplasmic detoxification |
| copF | 6119 | pMOL30 | cupA | 3524 | CHR1 | PIB1-type ATPase |
| silCBA | 6133–6136 | pMOL30 | cusCBA | 5031–5033 | CHR2 | HME-RND efflux pump |
| pTSS | sTSS | iTSS | aTSS | oTSS | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CT | Cu | ∩ | CT | Cu | ∩ | CT | Cu | ∩ | CT | Cu | ∩ | CT | Cu | ∩ | |
| CHR1 | 1490 | 1382 | 1231 | 729 | 734 | 483 | 1294 | 1390 | 916 | 1015 | 934 | 723 | 278 | 248 | 216 |
| CHR2 | 741 | 674 | 586 | 187 | 166 | 102 | 497 | 526 | 359 | 499 | 446 | 348 | 161 | 155 | 134 |
| pMOL28 | 45 | 43 | 38 | 19 | 19 | 13 | 89 | 83 | 74 | 97 | 92 | 77 | 21 | 22 | 18 |
| pMOL30 | 86 | 108 | 78 | 17 | 42 | 11 | 100 | 150 | 76 | 129 | 132 | 101 | 53 | 54 | 46 |
| Genome | 2362 | 2207 | 1933 | 952 | 961 | 609 | 1980 | 2149 | 1425 | 1740 | 1638 | 1249 | 513 | 479 | 414 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maertens, L.; Leys, N.; Matroule, J.-Y.; Van Houdt, R. The Transcriptomic Landscape of Cupriavidus metallidurans CH34 Acutely Exposed to Copper. Genes 2020, 11, 1049. https://doi.org/10.3390/genes11091049
Maertens L, Leys N, Matroule J-Y, Van Houdt R. The Transcriptomic Landscape of Cupriavidus metallidurans CH34 Acutely Exposed to Copper. Genes. 2020; 11(9):1049. https://doi.org/10.3390/genes11091049
Chicago/Turabian StyleMaertens, Laurens, Natalie Leys, Jean-Yves Matroule, and Rob Van Houdt. 2020. "The Transcriptomic Landscape of Cupriavidus metallidurans CH34 Acutely Exposed to Copper" Genes 11, no. 9: 1049. https://doi.org/10.3390/genes11091049
APA StyleMaertens, L., Leys, N., Matroule, J.-Y., & Van Houdt, R. (2020). The Transcriptomic Landscape of Cupriavidus metallidurans CH34 Acutely Exposed to Copper. Genes, 11(9), 1049. https://doi.org/10.3390/genes11091049