Clustered DNA Double-Strand Breaks: Biological Effects and Relevance to Cancer Radiotherapy

{kind=link}
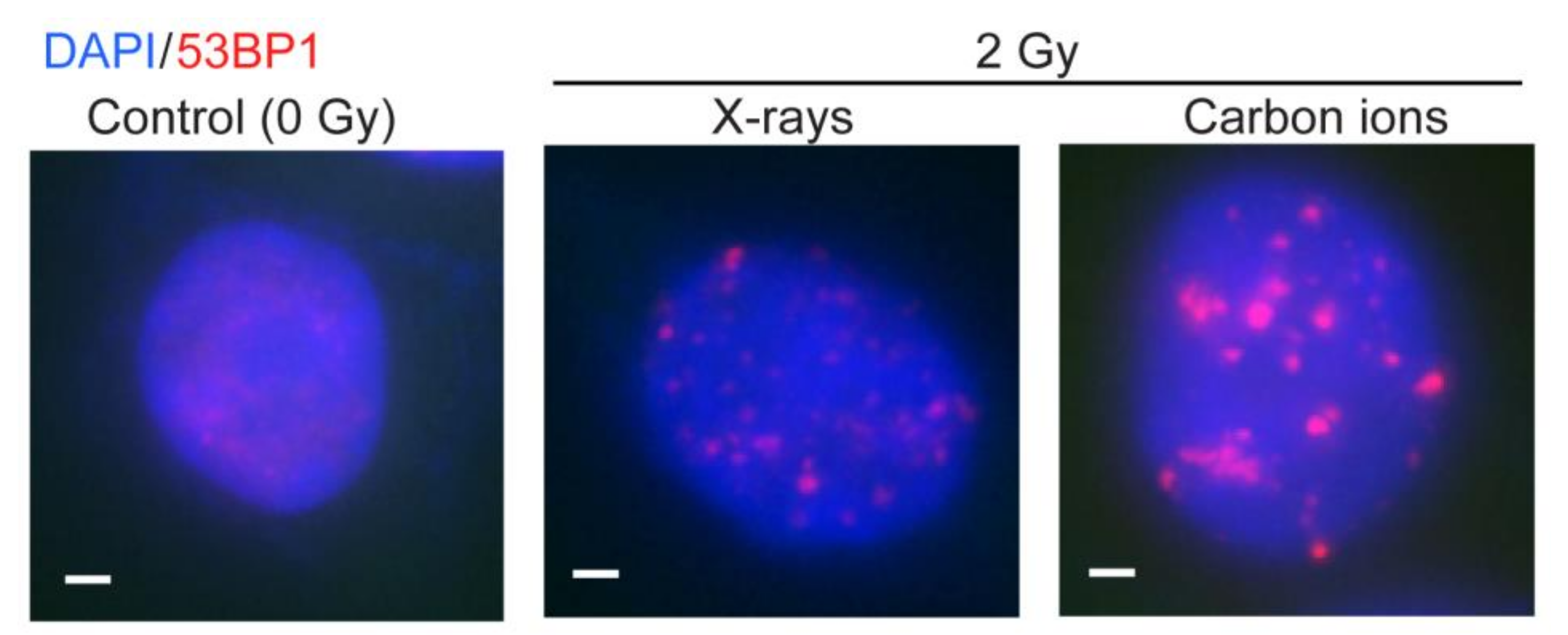{kind=link}
{kind=link}
{kind=link}
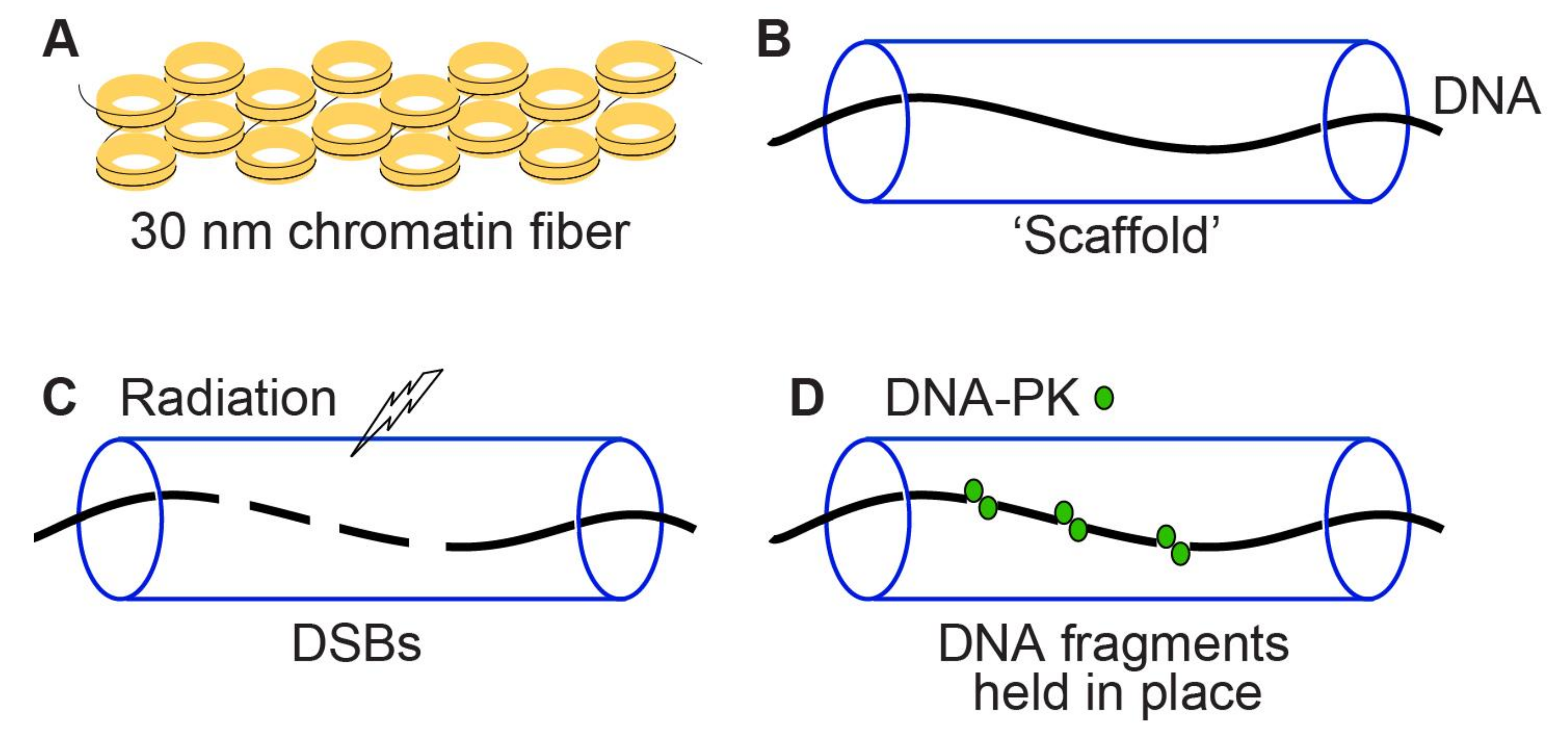{kind=link}
Abstract
1. Introduction
2. Radiation-Induced DNA Damage and the Importance of Clustered Lesions
3. Biological Effects of Clustered DNA Lesions
4. Low and High LET Radiation in Cancer Radiotherapy
5. Future Perspectives and Critical Remaining Questions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Bont, R.; Van Larebeke, N. Endogenous DNA Damage in Humans: A Review of Quantitative Data. Mutagen 2004, 19, 169–185. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Vilenchik, M.M.; Knudson, A.G. Endogenous DNA Double-Strand Breaks: Production, Fidelity of Repair, and Induction of Cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 12871–12876. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Haber, J.E. Sources of DNA Double-Strand Breaks and Models of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, M.C.; Noguchi, E. Regulation of DNA Replication through Natural Impediments in the Eukaryotic Genome. Genes 2017, 8, 98. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Ashley, A.K.; Hromas, R.; Nickoloff, J.A. More Forks on the Road to Replication Stress Recovery. J. Mol. Cell Biol. 2011, 3, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Budzowska, M.; Kanaar, R. Mechanisms of Dealing with DNA Damage-Induced Replication Problems. Cell Biochem. Biophys. 2009, 53, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and Consequences of Replication Stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Compe, E.; Egly, J.-M. Nucleotide Excision Repair and Transcriptional Regulation: TFIIH and Beyond. Annu. Rev. Biochem. 2016, 85, 265–290. [Google Scholar] [CrossRef]
- Mullins, E.A.; Rodriguez, A.A.; Bradley, N.P.; Eichman, B.F. Emerging Roles of DNA Glycosylases and the Base Excision Repair Pathway. Trends Biochem. Sci. 2019, 44, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.S. Base excision repair: A critical player in many games. DNA Repair 2014, 19, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Sage, E.; Harrison, L. Clustered DNA Lesion Repair in Eukaryotes: Relevance to Mutagenesis and Cell Survival. Mutat. Res. 2011, 711, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Sage, E.; Shikazono, N. Radiation-induced clustered DNA lesions: Repair and mutagenesis. Free Radic. Biol. Med. 2017, 107, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis, 2nd ed.; ASM Press: Washington, DC, USA, 2006. [Google Scholar]
- Rüthemann, P.; Pogliano, C.B.; Naegeli, H. Global-genome Nucleotide Excision Repair Controlled by Ubiquitin/Sumo Modifiers. Front. Genet. 2016, 7, 68. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Jones, D.; Lee, S.-H.; Williamson, E.A.; Hromas, R. Drugging the Cancers Addicted to DNA Repair. J. Natl. Cancer Inst. 2017, 109, djx059. [Google Scholar] [CrossRef]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA Double-Strand Break Repair Pathway Choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Budworth, H.; Matthewman, G.; O’Neill, P.; Dianov, G.L. Repair of Tandem Base Lesions in DNA by Human Cell Extracts Generates Persisting Single-strand Breaks. J. Mol. Biol. 2005, 351, 1020–1029. [Google Scholar] [CrossRef]
- Singleton, B.K.; Griffin, C.S.; Thacker, J. Clustered DNA damage leads to complex genetic changes in irradiated human cells. Cancer Res. 2002, 62, 6263–6269. [Google Scholar]
- Eccles, L.J.; O’Neill, P.; Lomax, M.E. Delayed Repair of Radiation Induced Clustered DNA Damage: Friend or Foe? Mutat. Res. 2011, 711, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Shikazono, N.; Noguchi, M.; Fujii, K.; Urushibara, A.; Yokoya, A. The yield, processing, and biological consequences of clustered DNA damage induced by ionizing radiation. J. Radiat. Res. 2009, 50, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Collins, L.B.; Chen, T.-H.; Herr, N.; Takeda, S.; Sun, W.; Swenberg, J.A.; Nakamura, J. Oxidative stress at low levels can induce clustered DNA lesions leading to NHEJ mediated mutations. Oncotarget 2016, 7, 25377–25390. [Google Scholar] [CrossRef]
- Magnander, K.; Elmroth, K. Biological consequences of formation and repair of complex DNA damage. Cancer Lett. 2012, 327, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G.; Mladenov, E.; Mladenova, V. Necessities in the Processing of DNA Double Strand Breaks and Their Effects on Genomic Instability and Cancer. Cancers 2019, 11, 1671. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Tomkinson, A.E. Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J. Biol. Chem. 2018, 293, 10536–10546. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G.; Murmann, T.; Soni, A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat. Res. Toxicol. Environ. Mutagen. 2015, 793, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Byrne, M.; Wray, J.; Reinert, B.; Wu, Y.; Nickoloff, J.; Lee, S.H.; Hromas, R.; Williamson, E. Mechanisms of Oncogenic Chromosomal Translocations. Ann. N. Y. Acad. Sci. 2014, 1310, 89–97. [Google Scholar] [CrossRef]
- Bunting, S.F.; Nussenzweig, A. End-joining, translocations and cancer. Nat. Rev. Cancer 2013, 13, 443–454. [Google Scholar] [CrossRef]
- Nickoloff, J.A. Recombination: Mechanisms and Roles in Tumorigenesis. In Encyclopedia of Cancer, 2nd ed.; Bertino, J.R., Ed.; Elsevier Science: San Diego, CA, USA, 2002; pp. 49–59. [Google Scholar]
- Nickoloff, J.A.; De Haro, L.P.; Wray, J.; Hromas, R. Mechanisms of leukemia translocations. Curr. Opin. Hematol. 2008, 15, 338–345. [Google Scholar] [CrossRef]
- Nickoloff, J.A. DNA Repair Dysregulation in Cancer: From Molecular Mechanisms to Synthetic Lethal Opportunities. In Stress Response Pathways in Cancer, from Molecular Targets to Novel Therapeutics; Wondrak, G.T., Ed.; Springer: New York, NY, USA, 2014; pp. 7–28. [Google Scholar]
- Symington, L.S.; Gautier, J. Double-Strand Break End Resection and Repair Pathway Choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Piazza, A.; Heyer, W.-D. Homologous Recombination and the Formation of Complex Genomic Rearrangements. Trends Cell Biol. 2019, 29, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.D.; Shah, S.S.; Heyer, W.-D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, J.A. Paths from DNA damage and signaling to genome rearrangements via homologous recombination. Mutat. Res. 2017, 806, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Semenenko, V.A.; Stewart, R.D. A fast Monte Carlo algorithm to simulate the spectrum of DNA damages formed by ionizing radiation. Radiat. Res. 2004, 161, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Goodhead, D.T. Initial Events in the Cellular Effects of Ionizing Radiations: Clustered Damage in DNA. Int. J. Radiat. Biol. 1994, 65, 7–17. [Google Scholar] [CrossRef]
- Goodhead, D.T.; Thacker, J.; Cox, R. Effects of Radiations of Different Qualities on Cells: Molecular Mechanisms of Damage and Repair. Int. J. Radiat. Biol. 1993, 63, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.N.S.U.W.E.W.M. Track structure in radiation biology: Theory and applications. Int. J. Radiat. Biol. 1998, 73, 355–364. [Google Scholar]
- Nikjoo, H.; O’Neill, P.; Goodhead, D.T.; Terrissol, M. Computational Modelling of Low-Energy Electron-Induced DNA Damage by Early Physical and Chemical Events. Int. J. Radiat. Biol. 1997, 71, 467–483. [Google Scholar] [CrossRef]
- Goodhead, D.T. Energy Deposition Stochastics and Track Structure: What About the Target? Radiat. Prot. Dosimetry 2006, 122, 3–15. [Google Scholar] [CrossRef]
- Nikjoo, H.; O’Neill, P.; Wilson, W.E.; Goodhead, D.T. Computational approach for determining the spectrum of DNA damage induced by ionizing radiation. Radiat. Res. 2001, 156 Pt 2, 577–583. [Google Scholar] [CrossRef]
- Dianov, G.L.; O’Neill, P.; Goodhead, D.T. Securing genome stability by orchestrating DNA repair: Removal of radiation-induced clustered lesions in DNA. BioEssays 2001, 23, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Goodhead, D.T. Mechanisms for the biological effectiveness of high-LET radiations. J. Radiat. Res. 1999, 40, S1–S13. [Google Scholar] [CrossRef] [PubMed]
- Nikjoo, H.; O’Neill, P.; Terrissol, M.; Goodhead, D.T. Quantitative modelling of DNA damage using Monte Carlo track structure method. Radiat. Environ. Biophys. 1999, 38, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Ward, J. Complexity of damage produced by ionizing radiation. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Lomax, M.; Folkes, L.; O’Neill, P. Biological Consequences of Radiation-induced DNA Damage: Relevance to Radiotherapy. Clin. Oncol. R. Coll. Radiol. 2013, 25, 578–585. [Google Scholar] [CrossRef]
- Harper, J.V.; Anderson, J.A.; O’Neill, P. Radiation induced DNA DSBs: Contribution from stalled replication forks? DNA Repair 2010, 9, 907–913. [Google Scholar] [CrossRef]
- Lomax, M.E.; Gulston, M.K.; O’Neil, P. Chemical Aspects of Clustered DNA Damage Induction by Ionising Radiation. Radiat. Prot. Dosim. 2002, 99, 63–68. [Google Scholar] [CrossRef]
- Cannan, W.J.; Tsang, B.P.; Wallace, S.S.; Pederson, D.S. Nucleosomes Suppress the Formation of Double-strand DNA Breaks during Attempted Base Excision Repair of Clustered Oxidative Damages. J. Biol. Chem. 2014, 289, 19881–19893. [Google Scholar] [CrossRef]
- Yang, N.; Galick, H.; Wallace, S.S. Attempted base excision repair of ionizing radiation damage in human lymphoblastoid cells produces lethal and mutagenic double strand breaks. DNA Repair 2004, 3, 1323–1334. [Google Scholar] [CrossRef]
- Blaisdell, J.O.; Harrison, L.; Wallace, S.S. Base excision repair processing of radiation-induced clustered DNA lesions. Radiat. Prot. Dosim. 2001, 97, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Ravanat, J.-L.; TavernaPorro, M.; Menoni, H.; Angelov, D. Oxidatively generated complex DNA damage: Tandem and clustered lesions. Cancer Lett. 2012, 327, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G.; O’Neill, P.; Stewart, R.D. Induction and Repair of Clustered DNA Lesions: What Do We Know So Far? Radiat. Res. 2013, 180, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Hada, M.; Georgakilas, A.G. Formation of clustered DNA damage after high-LET irradiation: A review. J. Radiat. Res. 2008, 49, 203–210. [Google Scholar] [CrossRef]
- Timm, S.; Lorat, Y.; Jakob, B.; Taucher-Scholz, G.; Rübe, C.E. Clustered DNA damage concentrated in particle trajectories causes persistent large-scale rearrangements in chromatin architecture. Radiother. Oncol. 2018, 129, 600–610. [Google Scholar] [CrossRef]
- Garty, G.; Schulte, R.; Shchemelinin, S.; Leloup, C.; Assaf, G.; Breskin, A.; Chechik, R.; Bashkirov, V.; Milligan, J.; Grosswendt, B. A nanodosimetric model of radiation-induced clustered DNA damage yields. Phys. Med. Biol. 2010, 55, 761–781. [Google Scholar] [CrossRef]
- Akamatsu, K.; Shikazono, N.; Saito, T. New method for estimating clustering of DNA lesions induced by physical/chemical mutagens using fluorescence anisotropy. Anal. Biochem. 2017, 536, 78–89. [Google Scholar] [CrossRef]
- Schuemann, J.; McNamara, A.L.; Warmenhoven, J.W.; Henthorn, N.T.; Kirkby, K.J.; Merchant, M.J.; Ingram, S.; Paganetti, H.; Held, K.D.; Ramos-Mendez, J.; et al. A New Standard DNA Damage (Sdd) Data Format. Radiat. Res. 2019, 191, 76–92. [Google Scholar] [CrossRef]
- Ward, J. The Nature of Lesions Formed by Ionizing Radiation. In DNA Damage and Repair: DNA Repair in Higher Eukaryotes; Nickoloff, J.A., Hoekstra, M.F., Eds.; Humana Press: Totowa, NJ, USA, 1998; pp. 65–84. [Google Scholar]
- Howard, M.; Beltran, C.; Sarkaria, J.; Herman, M.G. Characterization of relative biological effectiveness for conventional radiation therapy: A comparison of clinical 6 MV X-rays and 137Cs. J. Radiat. Res. 2017, 58, 608–613. [Google Scholar] [CrossRef]
- Asaithamby, A.; Chen, D.J. Cellular Responses to DNA Double-Strand Breaks after Low-Dose Γ-Irradiation. Nucleic Acids Res. 2009, 37, 3912–3923. [Google Scholar] [CrossRef]
- Brahme, A. Physical and Biologic Aspects on the Optimum Choice of Radiation Modality. Acta Radiol. Oncol. 1982, 21, 469–479. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tommasino, F.; Durante, M. Proton Radiobiology. Cancers 2015, 7, 353–381. [Google Scholar] [CrossRef] [PubMed]
- Paganetti, H. Relative biological effectiveness (RBE) values for proton beam therapy. Variations as a function of biological endpoint, dose, and linear energy transfer. Phys. Med. Biol. 2014, 59, R419–R472. [Google Scholar] [CrossRef] [PubMed]
- Mohamad, O.; Yamada, S.; Durante, M. Clinical Indications for Carbon Ion Radiotherapy. Clin. Oncol. 2018, 30, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, B.S.; Overgaard, J.; Bassler, N. In vitro RBE-LET dependence for multiple particle types. Acta Oncol. 2011, 50, 757–762. [Google Scholar] [CrossRef]
- Allen, C.; Borak, T.B.; Tsujii, H.; Nickoloff, J.A. Heavy charged particle radiobiology: Using enhanced biological effectiveness and improved beam focusing to advance cancer therapy. Mutat. Res. 2011, 711, 150–157. [Google Scholar] [CrossRef]
- Krämer, M. Treatment planning for heavy-ion radiotherapy: Biological optimization of multiple beam ports. J. Radiat. Res. 2001, 42, 39–46. [Google Scholar] [CrossRef][Green Version]
- Wedenberg, M.; Beltran, C.; Mairani, A.; Alber, M. Advanced Treatment Planning. Med Phys. 2018, 45, e1011–e1023. [Google Scholar] [CrossRef]
- Ward, J.F. DNA Damage Produced by Ionizing Radiation in Mammalian Cells: Identities, Mechanisms of Formation, and Reparability. Prog. Nucleic Acid Res. Mol. Biol. 1988, 35, 95–125. [Google Scholar]
- Budworth, H.; Dianov, G.L. Mode of Inhibition of Short-patch Base Excision Repair by Thymine Glycol within Clustered DNA Lesions. J. Biol. Chem. 2003, 278, 9378–9381. [Google Scholar] [CrossRef]
- Budworth, H.; Dianova, I.I.; Podust, V.N.; Dianov, G.L. Repair of Clustered DNA Lesions. Sequence-Specific Inhibition of Long-Patch Base Excision Repair Be 8-Oxoguanine. J. Biol. Chem. 2002, 277, 21300–21305. [Google Scholar] [CrossRef] [PubMed]
- Schipler, A.; Iliakis, G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013, 41, 7589–7605. [Google Scholar] [CrossRef] [PubMed]
- Lam, I.; Keeney, S. Mechanism and Regulation of Meiotic Recombination Initiation. Cold Spring Harb. Perspect. Biol. 2014, 7, a016634. [Google Scholar] [CrossRef] [PubMed]
- Stavnezer, J.; Guikema, J.E.; Schrader, C.E. Mechanism and regulation of class switch recombination. Annu. Rev. Immunol. 2008, 26, 261–292. [Google Scholar] [CrossRef] [PubMed]
- Roth, D.B. V(D)J Recombination: Mechanism, Errors, and Fidelity. Microbiol. Spectr. 2014, 2, 1–11. [Google Scholar] [CrossRef]
- Friedland, W.; Schmitt, E.; Kundrát, P.; Dingfelder, M.; Baiocco, G.; Barbieri, S.; Ottolenghi, A. Comprehensive track-structure based evaluation of DNA damage by light ions from radiotherapy-relevant energies down to stopping. Sci. Rep. 2017, 7, 45161. [Google Scholar] [CrossRef]
- McMahon, S.J.; Prise, K.M. Mechanistic Modelling of Radiation Responses. Cancers 2019, 11, 205. [Google Scholar] [CrossRef]
- Pang, D.; Chasovskikh, S.; Rodgers, J.E.; Dritschilo, A. Short DNA Fragments Are a Hallmark of Heavy Charged-Particle Irradiation and May Underlie Their Greater Therapeutic Efficacy. Front. Oncol. 2016, 6, 130. [Google Scholar] [CrossRef]
- Pang, D.; Rodgers, J.E.; Berman, B.L.; Chasovskikh, S.; Dritschilo, A. Spatial distribution of radiation-induced double-strand breaks in plasmid DNA as resolved by atomic force microscopy. Radiat. Res. 2005, 164, 755–765. [Google Scholar] [CrossRef]
- Okayasu, R. Repair of DNA Damage Induced by Accelerated Heavy Ions—A Mini Review. Int. J. Cancer 2012, 130, 991–1000. [Google Scholar] [CrossRef]
- Sutherland, B.M.; Bennett, P.V.; Schenk, H.; Sidorkina, O.; Laval, J.; Trunk, J.; Monteleone, D.; Sutherland, J. Clustered DNA damages induced by high and low LET radiation, including heavy ions. Phys. Medica 2001, 17, 202–204. [Google Scholar]
- Mladenov, E.; Saha, J.; Iliakis, G. Processing-Challenges Generated by Clusters of DNA Double-Strand Breaks Underpin Increased Effectiveness of High-LET Radiation and Chromothripsis. Neurotransm. Interact. Cogn. Funct. 2018, 1044, 149–168. [Google Scholar]
- Shibata, A. Regulation of repair pathway choice at two-ended DNA double-strand breaks. Mutat. Res. Mol. Mech. Mutagen. 2017, 803, 51–55. [Google Scholar] [CrossRef]
- Kakarougkas, A.; Jeggo, P.A. DNA DSB repair pathway choice: An orchestrated handover mechanism. Br. J. Radiol. 2014, 87, 20130685. [Google Scholar] [CrossRef] [PubMed]
- Durant, S.T.; Nickoloff, J.A. Good Timing in the Cell Cycle for Precise DNA Repair by BRCA1. Cell Cycle 2005, 4, 1216–1222. [Google Scholar] [CrossRef]
- Deriano, L.; Roth, D.B. Modernizing the Nonhomologous End-Joining Repertoire: Alternative and Classical NHEJ Share the Stage. Annu. Rev. Genet. 2013, 47, 433–455. [Google Scholar] [CrossRef] [PubMed]
- Pang, D.; Winters, T.A.; Jung, M.; Purkayastha, S.; Cavalli, L.R.; Chasovkikh, S.; Haddad, B.R.; Dritschilo, A. Radiation-generated short DNA fragments may perturb non-homologous end-joining and induce genomic instability. J. Radiat. Res. 2011, 52, 309–319. [Google Scholar] [CrossRef]
- Davis, A.J.; Chen, B.P.; Chen, D.J. DNA-PK: A dynamic enzyme in a versatile DSB repair pathway. DNA Repair 2014, 17, 21–29. [Google Scholar] [CrossRef]
- Kurimasa, A.; Kumano, S.; Boubnov, N.V.; Story, M.D.; Tung, C.-S.; Peterson, S.R.; Chen, D.J. Requirement for the Kinase Activity of Human DNA-Dependent Protein Kinase Catalytic Subunit in DNA Strand Break Rejoining. Mol. Cell. Biol. 1999, 19, 3877–3884. [Google Scholar] [CrossRef]
- Okayasu, R.; Okada, M.; Okabe, A.; Noguchi, M.; Takakura, K.; Takahashi, S. Repair of DNA damage induced by accelerated heavy ions in mammalian cells proficient and deficient in the non-homologous end-joining pathway. Radiat. Res. 2006, 165, 59–67. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, X.; Wang, P.; Yu, X.; Essers, J.; Chen, D.; Kanaar, R.; Takeda, S.; Wang, Y. Characteristics of DNA-binding proteins determine the biological sensitivity to high-linear energy transfer radiation. Nucleic Acids Res. 2010, 38, 3245–3251. [Google Scholar] [CrossRef] [PubMed]
- Hada, M.; Sutherland, B.M. Spectrum of complex DNA damages depends on the incident radiation. Radiat. Res. 2006, 165, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Kubo, M.; Ma, H.; Nakagawa, A.; Yoshida, Y.; Isono, M.; Kanai, T.; Ohno, T.; Furusawa, Y.; Funayama, T.; et al. Nonhomologous End-Joining Repair Plays a More Important Role than Homologous Recombination Repair in Defining Radiosensitivity after Exposure to High-LET Radiation. Radiat. Res. 2014, 182, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Yajima, H.; Fujisawa, H.; Nakajima, N.I.; Hirakawa, H.; Jeggo, P.A.; Okayasu, R.; Fujimori, A. The complexity of DNA double strand breaks is a critical factor enhancing end-resection. DNA Repair 2013, 12, 936–946. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Kurimasa, A.; Brenneman, M.A.; Chen, D.J.; Nickoloff, J.A. DNA-dependent protein kinase suppresses double-strand break-induced and spontaneous homologous recombination. Proc. Natl. Acad. Sci. USA 2002, 99, 3758–3763. [Google Scholar] [CrossRef]
- Allen, C.; Halbrook, J.; A Nickoloff, J. Interactive competition between homologous recombination and non-homologous end joining. Mol. Cancer Res. 2003, 1, 913–920. [Google Scholar]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef]
- Daley, J.M.; Sung, P. 53BP1, BRCA1, and the Choice between Recombination and End Joining at DNA Double-Strand Breaks. Mol. Cell. Biol. 2014, 34, 1380–1388. [Google Scholar] [CrossRef]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.-T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef]
- Fitzgerald, J.E.; Grenon, M.; Lowndes, N.F. 53BP1: Function and mechanisms of focal recruitment. Biochem. Soc. Trans. 2009, 37 Pt 1, 897–904. [Google Scholar] [CrossRef]
- Jakob, B.; Scholz, M.; Taucher-Scholz, G. Biological imaging of heavy charged-particle tracks. Radiat. Res. 2003, 159, 676–684. [Google Scholar] [CrossRef]
- Jakob, B.; Splinter, J.; Taucher-Scholz, G. Positional Stability of Damaged Chromatin Domains along Radiation Tracks in Mammalian Cells. Radiat. Res. 2009, 171, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.P.; Hirakawa, H.; Nakajima, N.I.; Moore, S.; Nie, J.; Sharma, N.; Sugiura, M.; Hoki, Y.; Araki, R.; Abe, M.; et al. Low- and High-LET Ionizing Radiation Induces Delayed Homologous Recombination that Persists for Two Weeks before Resolving. Radiat. Res. 2017, 188, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Penninckx, S.; Cekanaviciute, E.; Degorre, C.; Guiet, E.; Viger, L.; Lucas, S.; Costes, S.V.; Lucasb, S. Dose, LET and Strain Dependence of Radiation-Induced 53BP1 Foci in 15 Mouse Strains Ex Vivo Introducing Novel DNA Damage Metrics. Radiat. Res. 2019, 192, 1–12. [Google Scholar] [CrossRef]
- Jezkova, L.; Zadneprianetc, M.; Kulikova, E.; Smirnova, E.; Bulanova, T.; Depes, D.; Falkova, I.; Boreyko, A.; Krasavin, E.; Davidkova, M.; et al. Particles with Similar Let Values Generate DNA Breaks of Different Complexity and Reparability: A High-Resolution Microscopy Analysis of Yh2ax/53bp1 Foci. Nanoscale 2018, 10, 1162–1179. [Google Scholar] [CrossRef]
- Zhang, X.; Ye, C.; Sun, F.; Wei, W.; Hu, B.; Wang, J. Both Complexity and Location of DNA Damage Contribute to Cellular Senescence Induced by Ionizing Radiation. PLoS ONE 2016, 11, e0155725. [Google Scholar] [CrossRef]
- Schipler, A.; Mladenova, V.; Soni, A.; Nikolov, V.; Saha, J.; Mladenov, E.; Iliakis, G. Chromosome thripsis by DNA double strand break clusters causes enhanced cell lethality, chromosomal translocations and 53BP1-recruitment. Nucleic Acids Res. 2016, 44, 7673–7690. [Google Scholar] [CrossRef]
- Forment, J.V.; Kaidi, A.; Jackson, S.P. Chromothripsis and cancer: Causes and consequences of chromosome shattering. Nat. Rev. Cancer 2012, 12, 663–670. [Google Scholar] [CrossRef]
- Rode, A.; Maass, K.K.; Willmund, K.V.; Lichter, P.; Ernst, A. Chromothripsis in Cancer Cells: An Update. Int. J. Cancer 2016, 138, 2322–2333. [Google Scholar] [CrossRef]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event During Cancer Development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Tubio, J.M.; Estivill, X. Cancer: When Catastrophe Strikes a Cell. Nature 2011, 470, 476–477. [Google Scholar] [CrossRef] [PubMed]
- Ghezraoui, H.; Piganeau, M.; Renouf, B.; Renaud, J.-B.; Sallmyr, A.; Ruis, B.; Oh, S.; Tomkinson, A.E.; Hendrickson, E.A.; Giovannangeli, C.; et al. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol. Cell 2014, 55, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Soni, A.; Siemann, M.; Pantelias, G.E.; Iliakis, G. Marked contribution of alternative end-joining to chromosome-translocation-formation by stochastically induced DNA double-strand-breaks in G2-phase human cells. Mutat. Res. Toxicol. Environ. Mutagen. 2015, 793, 2–8. [Google Scholar] [CrossRef]
- Murshed, H. (Ed.) Fundamentals of Radiation Oncology, 3rd ed.; In Physical, Biological, and Clinical Aspects, Elsevier: Waltham, MA, USA, 2019. [Google Scholar]
- Wilson, R.R. Radiological Use of Fast Protons. Radiol. 1946, 47, 487–491. [Google Scholar] [CrossRef]
- Castro, J.R.; Quivey, J.M.; Lyman, J.T.; Chen, G.T.Y.; Phillips, T.L.; Tobias, C.A.; Alpen, E.L. Current status of clinical particle radiotherapy at Lawrence Berkeley laboratory. Cancer 1980, 46, 633–641. [Google Scholar] [CrossRef]
- Newhauser, W.D.; Durante, M. Assessing the risk of second malignancies after modern radiotherapy. Nat. Rev. Cancer 2011, 11, 438–448. [Google Scholar] [CrossRef]
- Tsujii, H.; Kamada, T. A review of update clinical results of carbon ion radiotherapy. Jpn. J. Clin. Oncol. 2012, 42, 670–685. [Google Scholar] [CrossRef]
- Tsujii, H.; Kamada, T.; Shirai, T.; Noda, K.; Tsuji, H.; Karasawa, K. (Eds.) Carbon-Ion Radiotherapy Principals, Practices, and Treatment Planning; Springer: Tokyo, Japan, 2014. [Google Scholar]
- Combs, S.E.; Kieser, M.; Rieken, S.; Habermehl, D.; Jäkel, O.; Haberer, T.; Nikoghosyan, A.; Haselmann, R.; Unterberg, A.; Wick, W.; et al. Randomized phase II study evaluating a carbon ion boost applied after combined radiochemotherapy with temozolomide versus a proton boost after radiochemotherapy with temozolomide in patients with primary glioblastoma: The CLEOPATRA Trial. BMC Cancer 2010, 10, 478. [Google Scholar] [CrossRef]
- Combs, S.E.; Burkholder, I.; Edler, L.; Rieken, S.; Habermehl, D.; Jäkel, O.; Haberer, T.; Haselmann, R.; Unterberg, A.; Wick, W.; et al. Randomised phase I/II study to evaluate carbon ion radiotherapy versus fractionated stereotactic radiotherapy in patients with recurrent or progressive gliomas: The CINDERELLA trial. BMC Cancer 2010, 10, 533. [Google Scholar] [CrossRef]
- Kong, L.; Gao, J.; Hu, J.; Lu, R.; Yang, J.; Qiu, X.; Hu, W.; Lu, J.J. Carbon ion radiotherapy boost in the treatment of glioblastoma: A randomized phase I/III clinical trial. Cancer Commun. 2019, 39, 5. [Google Scholar] [CrossRef]
- Nikoghosyan, A.V.; Karapanagiotou-Schenkel, I.; Munter, M.W.; Jensen, A.D.; Combs, S.E.; Debus, J. Randomised Trial of Proton Vs. Carbon Ion Radiation Therapy in Patients with Chordoma of the Skull Base, Clinical Phase Iii Study Hit-1-Study. BMC Cancer 2010, 10, 607. [Google Scholar] [CrossRef] [PubMed]
- Uhl, M.; Edler, L.; Jensen, A.D.; Habl, G.; Oelmann, J.; Röder, F.; Jäckel, O.; Debus, J.; Herfarth, K. Randomized phase II trial of hypofractionated proton versus carbon ion radiation therapy in patients with sacrococcygeal chordoma-the ISAC trial protocol. Radiat. Oncol. 2014, 9, 100. [Google Scholar] [CrossRef]
- Kamada, T.; Tsujii, H.; A Blakely, E.; Debus, J.; De Neve, W.; Durante, M.; Jäkel, O.; Mayer, R.; Orecchia, R.; Pötter, R.; et al. Carbon ion radiotherapy in Japan: An assessment of 20 years of clinical experience. Lancet Oncol. 2015, 16, e93–e100. [Google Scholar] [CrossRef]
- Miyamoto, T.; Baba, M.; Yamamoto, N.; Koto, M.; Sugawara, T.; Yashiro, T.; Kadono, K.; Ezawa, H.; Tsujii, H.; Mizoe, J.-E.; et al. Curative treatment of Stage I non–small-cell lung cancer with carbon ion beams using a hypofractionated regimen. Int. J. Radiat. Oncol. 2007, 67, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Suzuki, Y.; Ohno, T.; Kato, S.; Morita, S.; Sato, S.; Oka, K.; Tsujii, H. Carbon Beam Therapy Overcomes the Radiation Resistance of Uterine Cervical Cancer Originating from Hypoxia. Clin. Cancer Res. 2006, 12 Pt 1, 2185–2190. [Google Scholar] [CrossRef]
- Schulz-Ertner, D.; Karger, C.P.; Feuerhake, A.; Nikoghosyan, A.; Combs, S.E.; Jäkel, O.; Edler, L.; Scholz, M.; Debus, J. Effectiveness of Carbon Ion Radiotherapy in the Treatment of Skull-Base Chordomas. Int. J. Radiat. Oncol. 2007, 68, 449–457. [Google Scholar] [CrossRef]
- Murnane, J.P. Telomeres and chromosome instability. DNA Repair 2006, 5, 1082–1092. [Google Scholar] [CrossRef]
- Cannan, W.J.; Pederson, D.S. Chromosome Break. In Reference Module for Life Sciences; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- De Koning, A.P.J.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive Elements May Comprise Over Two-Thirds of the Human Genome. PLoS Genet. 2011, 7, e1002384. [Google Scholar] [CrossRef]
- Weinstock, D.M.; Brunet, E.; Jasin, M. Formation of Nhej-Derived Reciprocal Chromosomal Translocations Does Not Require Ku70. Nat. Cell Biol. 2007, 9, 978–981. [Google Scholar] [CrossRef]
- Zha, S.; Boboila, C.; Alt, F.W. Mre11: Roles in DNA repair beyond homologous recombination. Nat. Struct. Mol. Biol. 2009, 16, 798–800. [Google Scholar] [CrossRef]
- DeFazio, L.G.; Stansel, R.M.; Griffith, J.D.; Chu, G. Synapsis of DNA ends by DNA-dependent protein kinase. EMBO J. 2002, 21, 3192–3200. [Google Scholar] [CrossRef] [PubMed]
- Cornforth, M.; Bedford, J. X-ray–induced breakage and rejoining of human interphase chromosomes. Science 1983, 222, 1141–1143. [Google Scholar] [CrossRef] [PubMed]
- Joiner, M.; Bremner, J.; Denekamp, J.; Maughan, R. The Interaction between X-rays and 3 MeV Neutrons in the Skin of the Mouse Foot. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1984, 46, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Tilly, N. Comparison of cell survival models for mixed LET radiation. Int. J. Radiat. Biol. 1999, 75, 233–243. [Google Scholar] [CrossRef]
- Bird, R.P.; Zaider, M.; Rossi, H.H.; Rohrig, N.; Hall, E.J.; Rohrig, S.A.M. The Sequential Irradiation of Mammalian Cells with X Rays and Charged Particles of High LET. Radiat. Res. 1983, 93, 444–452. [Google Scholar] [CrossRef]
- Jensen, A.D.; Nikoghosyan, A.V.; Poulakis, M.; Höss, A.; Haberer, T.; Jäkel, O.; Münter, M.W.M.W.; Schulz-Ertner, D.; Huber, P.E.; Debus, J. Combined intensity-modulated radiotherapy plus raster-scanned carbon ion boost for advanced adenoid cystic carcinoma of the head and neck results in superior locoregional control and overall survival. Cancer 2015, 121, 3001–3009. [Google Scholar] [CrossRef]
- Combs, S.E.; Kessel, K.; Habermehl, D.; Haberer, T.; Jäkel, O.; Debus, J. Proton and carbon ion radiotherapy for primary brain tumors and tumors of the skull base. Acta Oncol. 2013, 52, 1504–1509. [Google Scholar] [CrossRef]
- Akbaba, S.; Held, T.; Lang, K.; Forster, T.; Federspil, P.; Herfarth, K.; Häfner, M.; Plinkert, P.; Rieken, S.; Debus, J.; et al. Bimodal Radiotherapy with Active Raster-Scanning Carbon Ion Radiotherapy and Intensity-Modulated Radiotherapy in High-Risk Nasopharyngeal Carcinoma Results in Excellent Local Control. Cancers 2019, 11, 379. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Kurka, T.; Jeggo, P.A. Kap-1 Phosphorylation Regulates Chd3 Nucleosome Remodeling During the DNA Double-Strand Break Response. Nat. Struct. Mol. Biol. 2010, 18, 831–839. [Google Scholar] [CrossRef]
- Arnoult, N.; Correia, A.; Ma, J.; Merlo, A.; Garcia-Gomez, S.; Maric, M.; Tognetti, M.; Benner, C.W.; Boulton, S.J.; Saghatelian, A.; et al. Regulation of DNA Repair Pathway Choice in S and G2 Phases by the Nhej Inhibitor Cyren. Nature 2017, 549, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Church, G.M. Simultaneous Discovery of Cell-Free DNA and the Nucleosome Ladder. Genetics 2018, 209, 27–29. [Google Scholar] [CrossRef]
- Flygare, J.; Armstrong, R.C.; Wennborg, A.; Orsan, S.; Hellgren, D. Proteolytic cleavage of HsRad51 during apoptosis. FEBS Lett. 1998, 427, 247–251. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nickoloff, J.A.; Sharma, N.; Taylor, L. Clustered DNA Double-Strand Breaks: Biological Effects and Relevance to Cancer Radiotherapy. Genes 2020, 11, 99. https://doi.org/10.3390/genes11010099
Nickoloff JA, Sharma N, Taylor L. Clustered DNA Double-Strand Breaks: Biological Effects and Relevance to Cancer Radiotherapy. Genes. 2020; 11(1):99. https://doi.org/10.3390/genes11010099
Chicago/Turabian StyleNickoloff, Jac A., Neelam Sharma, and Lynn Taylor. 2020. "Clustered DNA Double-Strand Breaks: Biological Effects and Relevance to Cancer Radiotherapy" Genes 11, no. 1: 99. https://doi.org/10.3390/genes11010099
APA StyleNickoloff, J. A., Sharma, N., & Taylor, L. (2020). Clustered DNA Double-Strand Breaks: Biological Effects and Relevance to Cancer Radiotherapy. Genes, 11(1), 99. https://doi.org/10.3390/genes11010099