Rewriting Human History and Empowering Indigenous Communities with Genome Editing Tools


{kind=link}
{kind=link}
Abstract
1. Introduction
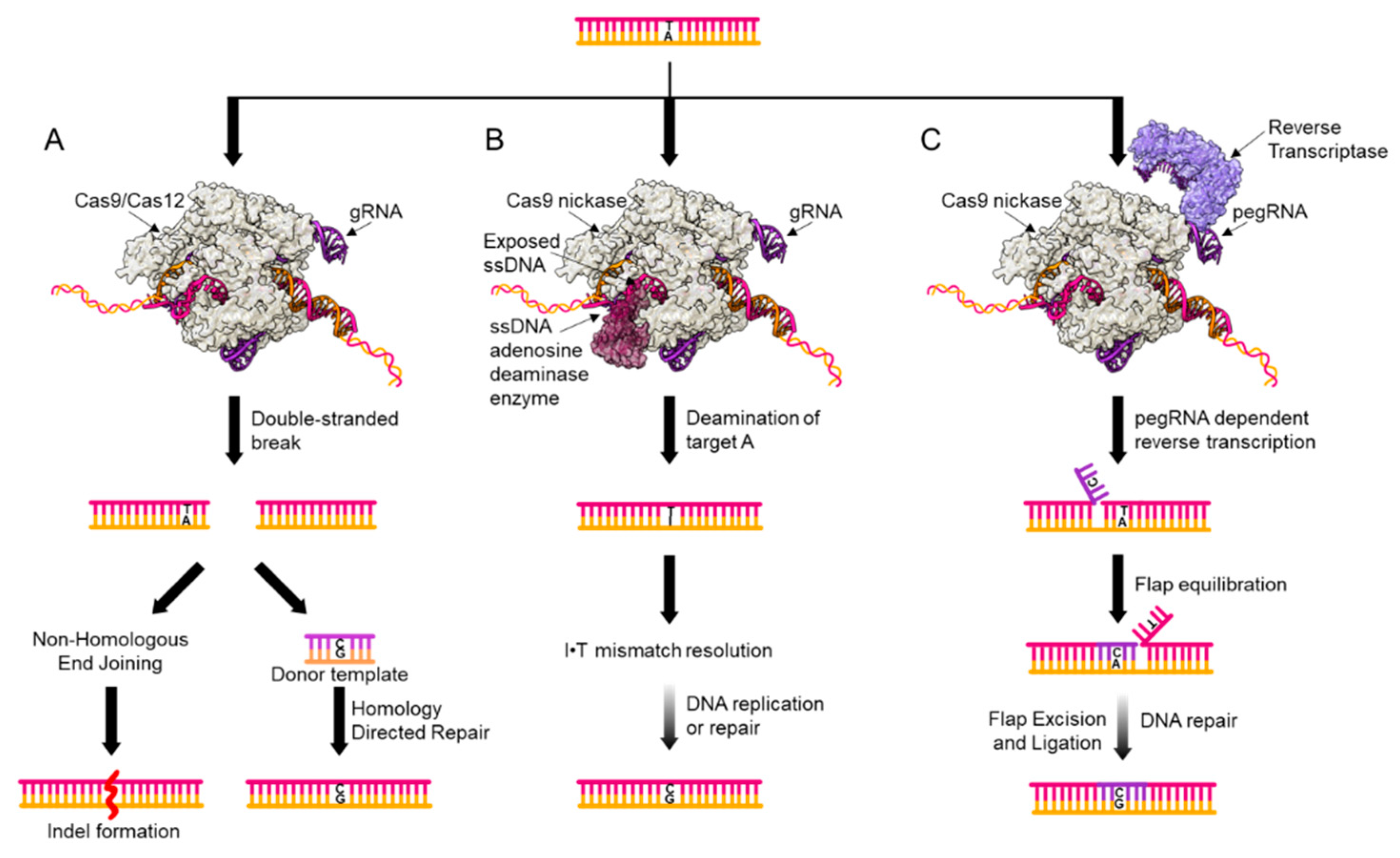
2. Genome Editing Tools for Point Mutation Introduction
3. Limitations of Current Tools
4. Signatures of Natural Selection
5. The “Thrifty Gene Hypothesis”
6. Previous Methods for Functional Investigation
7. Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Starita, L.M.; David, L.; Young, M.I.; Jacob, O.; Kitzman, J.G.; Ronald, J.; Hause, D.M.; Fowler, J.D.; Parvin, J.S.; Fields, S. Massively Parallel Functional Analysis of BRCA1 RING Domain Variants. Genetics 2015, 200, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Nishimasu, H.; Shi, X.; Ishiguro, S.; Gao, L.; Hirano, S.; Okazaki, S.; Noda, T.; Abudayyeh, O.O.; Gootenberg, J.S.; Mori, H.; et al. Engineered CRISPR-Cas9 Nuclease with Expanded Targeting Space. Science 2018, 361, 1259–1262. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Topkar, V.V.; Zheng, Z.; Joung, J.K. Broadening the Targeting Range of Staphylococcus Aureus CRISPR-Cas9 by Modifying PAM Recognition. Nat. Biotechnol. 2015, 33, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Slaymaker, I.M.; Linyi Gao, B.Z.; Scott, W.; Zhang, F. Rationally Engineered Cas9 Nucleases with Improved Specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-Fidelity CRISPR-Cas9 Nucleases with No Detectable Genome-Wide off-Target Effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Franklin, B.; Koob, J.; Kellner, M.J.; Ladha, A.; Joung, J.; Kirchgatterer, P.; Cox, D.B.; Zhang, F. A Cytosine Deaminase for Programmable Single-Base RNA Editing. Science 2019, 365, 382–386. [Google Scholar] [CrossRef]
- Cox, D.B.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA Editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable Base Editing of A T to G C in Genomic DNA without DNA Cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable Editing of a Target Base in Genomic DNA without Double-Stranded DNA Cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Nishida, K.; Arazoe, T.; Yachie, N.; Banno, S.; Kakimoto, M.; Tabata, M.; Mochizuki, M.; Miyabe, A.; Araki, M.; Hara, K.Y.; et al. Targeted Nucleotide Editing Using Hybrid Prokaryotic and Vertebrate Adaptive Immune Systems. Science 2016, 353. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-Replace Genome Editing without Double-Strand Breaks or Donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Hansen, M.E.; Lo, Y.; Tishkoff, S.A. Going Global by Adapting Local: A Review of Recent Human Adaptation. Science 2016, 354, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Castanon, O.; Said, K.; Volf, V.; Khoshakhlagh, P.; Hornick, A.; Ferreira, R.; Wu, C.T.; Güell, M.; Garg, S.; et al. Enabling Large-Scale Genome Editing by Reducing DNA Nicking. BioRxiv 2019, 574020. [Google Scholar] [CrossRef]
- Neel, J.V. Diabetes Mellitus: A ‘Thrifty’ Genotype Rendered Detrimental by Progress? Am. J. Hum. Genet. 1962, 14, 353–362. [Google Scholar]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef]
- Jiang, F.; Taylor, D.W.; Chen, J.S.; Kornfeld, J.E.; Zhou, K.; Thompson, A.J.; Nogales, E.; Doudna, J.A. Structures of a CRISPR-Cas9 R-Loop Complex Primed for DNA Cleavage. Science 2016, 351, 867–871. [Google Scholar] [CrossRef]
- Jeggo, P.A. DNA Breakage and Repair. Adv. Genet. 1998, 38, 185–218. [Google Scholar] [CrossRef]
- Rouet, P.; Smih, F.; Jasin, M. Introduction of Double-Strand Breaks into the Genome of Mouse Cells by Expression of a Rare-Cutting Endonuclease. Mol. Cell. Biol. 1994, 14, 8096–8106. [Google Scholar] [CrossRef]
- Rudin, N.; Sugarman, E.; Haber, J.E. Genetic and Physical Analysis of Double-Strand Break Repair and Recombination in Saccharomyces Cerevisiae. Genetics 1989, 122, 519–534. [Google Scholar]
- Jasin, M. Genetic Manipulation of Genomes with Rare-Cutting Endonucleases. Trends Genet. 1996, 12, 224–228. [Google Scholar] [CrossRef]
- Li, X.; Wang, Y.; Liu, Y.; Yang, B.; Wang, X.; Wei, J.; Lu, Z.; Zhang, Y.; Wu, J.; Huang, X.; et al. Base Editing with a Cpf1-Cytidine Deaminase Fusion. Nat. Biotechnol. 2018, 36, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Zhao, K.T.; Packer, M.S.; Gaudelli, N.M.; Waterbury, A.L.; Koblan, L.W.; Kim, Y.B.; Badran, A.H.; Liu, D.R. Improved Base Excision Repair Inhibition and Bacteriophage Mu Gam Protein Yields C:G-to-T:A Base Editors with Higher Efficiency and Product Purity. Sci. Adv. 2017, 3, eaao4774. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Feng, S.; Huang, S.; Yu, W.; Li, G.; Yang, G.; Liu, Y.; Zhang, Y.; Zhang, L.; Hou, Y.; et al. BE-PLUS: A New Base Editing Tool with Broadened Editing Window and Enhanced Fidelity. Cell Res. 2018, 28, 855–861. [Google Scholar] [CrossRef]
- Kim, Y.B.; Komor, A.C.; Levy, J.M.; Packer, M.S.; Zhao, K.T.; Liu, D.R. Increasing the Genome-Targeting Scope and Precision of Base Editing with Engineered Cas9-Cytidine Deaminase Fusions. Nat. Biotechnol. 2017, 35, 371–376. [Google Scholar] [CrossRef]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 Variants with Broad PAM Compatibility and High DNA Specificity. Nature 2018, 556, 57–63. [Google Scholar] [CrossRef]
- Endo, M.; Mikami, M.; Endo, A.; Kaya, H.; Itoh, T.; Nishimasu, H.; Nureki, O.; Toki, S. Genome Editing in Plants by Engineered CRISPR-Cas9 Recognizing NG PAM. Nat. Plants 2019, 5, 14–17. [Google Scholar] [CrossRef]
- Rees, H.A.; Komor, A.C.; Yeh, W.H.; Caetano-Lopes, J.; Warman, M.; Edge, A.S.; Liu, D.R. Improving the DNA Specificity and Applicability of Base Editing through Protein Engineering and Protein Delivery. Nat. Commun. 2017, 8, 15790. [Google Scholar] [CrossRef]
- Liang, P.; Ding, C.; Sun, H.; Xie, X.; Xu, Y.; Zhang, X.; Sun, Y.; Xiong, Y.; Ma, W.; Liu, Y.; et al. Correction of β-Thalassemia Mutant by Base Editor in Human Embryos. Protein Cell 2017, 8, 811–822. [Google Scholar] [CrossRef]
- Ryu, S.M.; Koo, T.; Kim, K.; Lim, K.; Baek, G.; Kim, S.; Kim, H.S.; Kim, D.E.; Lee, H.; Chung, E.; et al. Adenine Base Editing in Mouse Embryos and an Adult Mouse Model of Duchenne Muscular Dystrophy. Nat. Biotechnol. 2018, 36, 536–539. [Google Scholar] [CrossRef]
- Yeh, W.H.; Chiang, H.; Rees, H.A.; Edge, A.S.; Liu, D.R. In Vivo Base Editing of Post-Mitotic Sensory Cells. Nat. Commun. 2018, 9, 2184. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.; Sun, H.; Sun, Y.; Zhang, X.; Xie, X.; Zhang, J.; Zhang, Z.; Chen, Y.; Ding, C.; Xiong, Y.; et al. Effective Gene Editing by High-Fidelity Base Editor 2 in Mouse Zygotes. Protein Cell 2017, 8, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, M.; Chen, S.; Deng, J.; Song, Y.; Lai, L.; Li, Z. Highly Efficient RNA-Guided Base Editing in Rabbit. Nat. Commun. 2018, 9, 2717. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yu, L.; Zhang, X.; Xin, C.; Huang, S.; Bai, L.; Chen, W.; Gao, R.; Li, J.; Pan, S.; et al. Highly Efficient and Precise Base Editing by Engineered DCas9-Guide TRNA Adenosine Deaminase in Rats. Cell Discov. 2018, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.D.; Joung, J.K. CRISPR-Cas Systems for Editing, Regulating and Targeting Genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Saleh-Gohari, N.; Helleday, T. Conservative Homologous Recombination Preferentially Repairs DNA Double-Strand Breaks in the S Phase of the Cell Cycle in Human Cells. Nucleic Acids Res. 2004, 32, 3683–3688. [Google Scholar] [CrossRef]
- Heyer, W.D.; Ehmsen, K.T.; Liu, J. Regulation of Homologous Recombination in Eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR-Cas9 Genome Editing Induces a P53-Mediated DNA Damage Response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. P53 Inhibits CRISPR-Cas9 Engineering in Human Pluripotent Stem Cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef]
- Webber, B.R.; Lonetree, C.L.; Kluesner, M.G.; Johnson, M.J.; Pomeroy, E.J.; Diers, M.D.; Lahr, W.S.; Draper, G.M.; Slipek, N.J.; Smeester, B.S.; et al. Highly Efficient Multiplex Human T Cell Engineering without Double-Strand Breaks Using Cas9 Base Editors. Nat. Commun. 2019, 10, 5222. [Google Scholar] [CrossRef]
- Kim, D.; Lim, K.; Kim, S.T.; Yoon, S.H.; Kim, K.; Ryu, S.M.; Kim, J.S. Genome-Wide Target Specificities of CRISPR RNA-Guided Programmable Deaminases. Nat. Biotechnol. 2017, 35, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, D.E.; Lee, G.; Cho, S.I.; Kim, J.S. Genome-Wide Target Specificity of CRISPR RNA-Guided Adenine Base Editors. Nat. Biotechnol. 2019, 37, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, J.M.; Cervantes, O.; Clement, M.K.; Wu, Y.; Zeng, J.; Bauer, D.E.; Pinello, L.; Joung, J.K. An APOBEC3A-Cas9 Base Editor with Minimized Bystander and off-Target Activities. Nat. Biotechnol. 2018, 36, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Zong, Y.; Gao, Q.; Zhu, Z.; Wang, Y.; Qin, P.; Liang, C.; Wang, D.; Qiu, J.L.; Zhang, F.; et al. Cytosine, but Not Adenine, Base Editors Induce Genome-Wide off-Target Mutations in Rice. Science 2019, 364, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Zuo, E.; Sun, Y.; Wei, W.; Yuan, T.; Ying, W.; Sun, H.; Yuan, L.; Steinmetz, L.M.; Li, Y.; Yang, H. Cytosine Base Editor Generates Substantial Off-Target Single-Nucleotide Variants in Mouse Embryos. Science 2019, 364, 289–292. [Google Scholar] [CrossRef]
- Grünewald, J.; Zhou, R.; Iyer, S.; Lareau, C.A.; Garcia, S.P.; Aryee, M.J.; Joung, J.K. CRISPR DNA Base Editors with Reduced RNA Off-Target and Self-Editing Activities. Nat. Biotechnol. 2019, 37, 1041–1048. [Google Scholar] [CrossRef]
- Grünewald, J.; Zhou, R.; Garcia, S.P.; Iyer, S.; Lareau, C.A.; Aryee, M.J.; Joung, J.K. Transcriptome-Wide off-Target RNA Editing Induced by CRISPR-Guided DNA Base Editors. Nature 2019, 569, 433–437. [Google Scholar] [CrossRef]
- Zhou, C.; Sun, Y.; Yan, R.; Liu, Y.; Zuo, E.; Gu, C.; Han, L.; Wei, Y.; Hu, X.; Zeng, R.; et al. Off-Target RNA Mutation Induced by DNA Base Editing and Its Elimination by Mutagenesis. Nature 2019, 571, 275–278. [Google Scholar] [CrossRef]
- Moreno-Mayar, J.V.; Vinner, L.; De Barros Damgaard, P.; De La Fuente, C.; Chan, J.; Spence, J.P.; Allentoft, M.E.; Vimala, T.; Racimo, F.; Pinotti, T.; et al. Early Human Dispersals within the Americas. Science 2018, 362. [Google Scholar] [CrossRef]
- Knapp, M.; Horsburgh, K.A.; Prost, S.; Stanton, J.A.; Buckley, H.R.; Walter, R.K.; Matisoo-Smith, E.A. Complete Mitochondrial DNA Genome Sequences from the First New Zealanders. Proc. Natl. Acad. Sci. USA 2012, 109, 18350–18354. [Google Scholar] [CrossRef]
- Dannemann, M.; Janet, K. The Contribution of Neanderthals to Phenotypic Variation in Modern Humans. Am. J. Hum. Genet. 2017, 101, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.C. Protection Afforded by Sickle–Cell Trait against Subtertian Malarial Infection. Br. Med. J. 1954, 1, 290–294. [Google Scholar] [CrossRef]
- Thom, C.S.; Dickson, C.F.; Gell, D.A.; Weiss, M.J. Hemoglobin Variants: Biochemical Properties and Clinical Correlates. Cold Spring Harb. Perspect. Med. 2013, 3, a011858. [Google Scholar] [CrossRef] [PubMed]
- Luzzatto, L. Sickle Cell Anaemia and Malaria. Mediterr. J. Hematol. Infect. Dis. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, F.R.; Huff, C.; Myllymäki, M.; Olenchock, B.; Swierczek, S.; Tashi, T.; Gordeuk, V.; Wuren, T.; Ri-Li, G.; McClain, D.A.; et al. A Genetic Mechanism for Tibetan High-Altitude Adaptation. Nat. Genet. 2014, 46, 951–956. [Google Scholar] [CrossRef]
- Fumagalli, M.; Moltke, I.; Grarup, N.; Racimo, F.; Bjerregaard, P.; Jørgensen, M.E.; Korneliussen, T.S.; Gerbault, P.; Skotte, L.; Linneberg, A.; et al. Greenlandic Inuit Show Genetic Signatures of Diet and Climate Adaptation. Science 2015, 349, 1343–1347. [Google Scholar] [CrossRef]
- Ilardo, M.A.; Moltke, I.; Korneliussen, T.S.; Cheng, J.; Stern, A.J.; Racimo, F.; De Barros Damgaard, P.; Sikora, M.; Seguin-Orlando, A.; Rasmussen, S.; et al. Physiological and Genetic Adaptations to Diving in Sea Nomads. Cell 2018, 173, 569–580. [Google Scholar] [CrossRef]
- Bigham, A.W.; Wilson, M.J.; Julian, C.G.; Kiyamu, M.; Vargas, E.; Leon-Velarde, F.; Rivera-Chira, M.; Rodriquez, C.; Browne, V.A.; Parra, E.; et al. Andean and Tibetan Patterns of Adaptation to High Altitude. Am. J. Hum. Biol. Off. J. Hum. Biol. Counc. 2013, 25, 190–197. [Google Scholar] [CrossRef]
- Tishkoff, S. Strength in Small Numbers. Science 2015, 349, 1282–1283. [Google Scholar] [CrossRef]
- Mills, M.C.; Charles, R. A Scientometric Review of Genome-Wide Association Studies. Commun. Biol. 2019, 2, 1–11. [Google Scholar] [CrossRef]
- Gould, S.J.; Lewontin, R.C. The Spandrels of San Marco and the Panglossian Paradigm: A Critique of the Adaptationist Programme. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1979, 205, 581–598. [Google Scholar] [CrossRef]
- Minster, R.L.; Hawley, N.L.; Su, C.T.; Sun, G.; Kershaw, E.E.; Cheng, H.; Buhule, O.D.; Lin, J.; Tuitele, J.; Naseri, T.; et al. A Thrifty Variant in CREBRF Strongly Influences Body Mass Index in Samoans. Nat. Genet. 2016, 48, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Loos, R.J.F. CREBRF Variant Increases Obesity Risk and Protects against Diabetes in Samoans. Nat. Genet. 2016, 48, 976–978. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, M.; Major, T.J.; Topless, R.K.; Dewes, O.; Yu, L.; Thompson, J.M.; McCowan, L.; De Zoysa, J.; Stamp, L.K.; Dalbeth, N.; et al. Discordant Association of the CREBRF Rs373863828 A Allele with Increased BMI and Protection from Type 2 Diabetes in Māori and Pacific (Polynesian) People Living in Aotearoa/New Zealand. Diabetologia 2018, 61, 1603–1613. [Google Scholar] [CrossRef]
- Hall, C.L.; Sutanto, H.; Dalageorgou, C.; McKenna, W.J.; Syrris, P.; Futema, M. Frequency of Genetic Variants Associated with Arrhythmogenic Right Ventricular Cardiomyopathy in the Genome Aggregation Database. Eur. J. Hum. Genet. 2018, 26, 1312–1318. [Google Scholar] [CrossRef]
- Claussnitzer, M.; Dankel, S.N.; Kim, K.H.; Quon, G.; Meuleman, W.; Haugen, C.; Glunk, V.; Sousa, I.S.; Beaudry, J.L.; Puviindran, V.; et al. FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N. Engl. J. Med. 2015, 373, 895–907. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Mark, A.H. Living with the Past: Evolution, Development, and Patterns of Disease. Science 2004, 305, 1733–1736. [Google Scholar] [CrossRef]
- Berry, S.D.; Walker, C.G.; Ly, K.; Snell, R.G.; Carr, P.A.; Bandara, D.; Mohal, J.; Castro, T.G.; Marks, E.J.; Morton, S.M.B.; et al. Widespread Prevalence of a CREBRF Variant amongst Māori and Pacific Children Is Associated with Weight and Height in Early Childhood. Int. J. Obes. 2018, 42, 603. [Google Scholar] [CrossRef]
- Gosling, A.L.; Buckley, H.R.; Matisoo-Smith, E.; Merriman, T.R. Pacific Populations, Metabolic Disease and Just-So Stories: A Critique of the Thrifty Genotype Hypothesis in Oceania. Ann. Hum. Genet. 2015, 79, 470–480. [Google Scholar] [CrossRef]
- Bird, D.W.; Rebecca, L.; Bliege, B. The Science of Foragers: Evaluating Variability among Hunter-Gatherers. Antiquity 1997, 71, 477–480. [Google Scholar] [CrossRef]
- Bellwood, P. Early Agriculturalist Population Diasporas? Farming, Languages, and Genes. Annu. Rev. Anthropol. 2001, 30, 181–207. [Google Scholar] [CrossRef]
- Stipp, D. Linking Nutrition, Maturation and Aging: From Thrifty Genes to the Spendthrift Phenotype. Aging 2011, 3, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Benyshek, D.C.; James, T.W. Exploring the Thrifty Genotype’s Food-Shortage Assumptions: A Cross-Cultural Comparison of Ethnographic Accounts of Food Security among Foraging and Agricultural Societies. Am. J. Phys. Anthropol. 2006, 131, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Speakman, J.R. Thrifty Genes for Obesity and the Metabolic Syndrome—Time to Call off the Search? Diabetes Vasc. Dis. Res. 2006, 3, 7–11. [Google Scholar] [CrossRef]
- Paradies, Y.C.; Michael, J.M.; Stephanie, M.F. Racialized Genetics and the Study of Complex Diseases: The Thrifty Genotype Revisited. Perspect. Biol. Med. 2007, 50, 203–227. [Google Scholar] [CrossRef]
- Nielsen, R. Adaptionism-30 Years after Gould and Lewontin. Evol. Int. J. Org. Evol. 2009, 63, 2487–2490. [Google Scholar] [CrossRef]
- Mulrooney, M.A.; Thegn, N.L. Hawaiian heiau and agricultural production in the kohala dryland field system. J. Polyn. Soc. 2005, 114, 45–67. [Google Scholar]
- Roullier, C.; Benoit, L.; McKey, D.B.; Lebot, V. Historical Collections Reveal Patterns of Diffusion of Sweet Potato in Oceania Obscured by Modern Plant Movements and Recombination. Proc. Natl. Acad. Sci. USA 2013, 110, 2205–2210. [Google Scholar] [CrossRef]
- Finney, B.R. Voyaging Canoes and the Settlement of Polynesia. Science 1977, 196, 1277–1285. [Google Scholar] [CrossRef]
- Zimmet, P.; Alberti, K.G.; Shaw, J. Global and Societal Implications of the Diabetes Epidemic. Nature 2001, 414, 782–787. [Google Scholar] [CrossRef]
- Lindo, J.; Huerta-Sánchez, E.; Nakagome, S.; Rasmussen, M.; Petzelt, B.; Mitchell, J.; Cybulski, J.S.; Willerslev, E.; DeGiorgio, M.; Malhi, R.S. A Time Transect of Exomes from a Native American Population before and after European Contact. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R. New Therapeutic Approaches for the Treatment of Obesity. Sci. Transl. Med. 2016, 8, 323rv2. [Google Scholar] [CrossRef] [PubMed]
- Reilly, S.M.; Alan, R.S. Adapting to Obesity with Adipose Tissue Inflammation. Nat. Rev. Endocrinol. 2017, 13, 633–643. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, D.G.; Manolio, T.A.; Dimmock, D.P.; Rehm, H.L.; Shendure, J.; Abecasis, G.R.; Adams, D.R.; Altman, R.B.; Antonarakis, S.E.; Ashley, E.A.; et al. Guidelines for Investigating Causality of Sequence Variants in Human Disease. Nature 2014, 508, 469–476. [Google Scholar] [CrossRef]
- Martin, A.R.; Kanai, M.; Kamatani, Y.; Okada, Y.; Benjamin, N.; Mark, M.; Daly, J. Clinical Use of Current Polygenic Risk Scores May Exacerbate Health Disparities. Nat. Genet. 2019, 51, 584–591. [Google Scholar] [CrossRef]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and Intensity of Constraint in Mammalian Genomic Sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A General Framework for Estimating the Relative Pathogenicity of Human Genetic Variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Starita, L.M.; Nadav, A.M.; Dunham, J.; Kitzman, F.; Roth, G.S.; Douglas, M.F. Variant Interpretation, Functional Assays to the Rescue. Am. J. Hum. Genet. 2017, 101, 315–325. [Google Scholar] [CrossRef]
- Chiasson, M.; Douglas, M.F. Mutagenesis-Based Protein Structure Determination. Nat. Genet. 2019, 51, 1072–1073. [Google Scholar] [CrossRef]
- Hasle, N.; Cooke, A.; Srivatsan, S.; Huang, H.; Stephany, J.J.; Krieger, Z.; Jackson, D.L.; Tang, W.; Monnat, R.J.; Trapnell, C.; et al. Visual Cell Sorting: A High-Throughput, Microscope-Based Method to Dissect Cellular Heterogeneity. BioRxiv 2019, 856476. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the CRISPR-Cas9 System. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Standage-Beier, K.; Tekel, S.J.; Brookhouser, N.; Schwarz, G.; Nguyen, T.; Wang, X.; Brafman, D.A. A Transient Reporter for Editing Enrichment (TREE) in Human Cells. Nucleic Acids Res. 2019, 47, e120. [Google Scholar] [CrossRef] [PubMed]
- Bigham, A.W.; Frank, S.L. Human High-Altitude Adaptation: Forward Genetics Meets the HIF Pathway. Genes Dev. 2014, 28, 2189–2204. [Google Scholar] [CrossRef] [PubMed]
- Kasendra, M.; Tovaglieri, A.; Sontheimer-Phelps, A.; Jalili-Firoozinezhad, S.; Bein, A.; Chalkiadaki, A.; Scholl, W.; Zhang, C.; Rickner, H.; Richmond, C.A.; et al. Development of a Primary Human Small Intestine-on-a-Chip Using Biopsy-Derived Organoids. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Claw, K.G.; Anderson, M.Z.; Begay, R.L.; Tsosie, K.S.; Fox, K.; Garrison, N.A. Summer internship for INdigenous peoples in Genomics (SING) Consortium. A framework for enhancing ethical genomic research with Indigenous communities. Nat. Commun. 2018, 9, 2957. [Google Scholar] [CrossRef]
- Vickers, S.M.; Fouad, M.N. An overview of EMPaCT and fundamental issues affecting minority participation in cancer clinical trials: Enhancing minority participation in clinical trials (EMPaCT): Laying the groundwork for improving minority clinical trial accrual. Cancer 2014, 120 (Suppl. 7), 1087–1090. [Google Scholar] [CrossRef]
- Steinthorsdottir, V.; Thorleifsson, G.; Sulem, P.; Helgason, H.; Grarup, N.; Sigurdsson, A.; Helgadottir, H.T.; Johannsdottir, H.; Magnusson, O.T.; Gudjonsson, S.A.; et al. Identification of Low-Frequency and Rare Sequence Variants Associated with Elevated or Reduced Risk of Type 2 Diabetes. Nat. Genet. 2014, 46, 294–298. [Google Scholar] [CrossRef]
- Dever, D.P.; Bak, R.O.; Reinisch, A.; Camarena, J.; Washington, G.; Nicolas, C.E.; Pavel-Dinu, M.; Saxena, N.; Wilkens, A.B.; Mantri, S.; et al. CRISPR/Cas9 β-Globin Gene Targeting in Human Haematopoietic Stem Cells. Nature 2016, 539, 384–389. [Google Scholar] [CrossRef]
- King, M.C.; Marks, J.H.; Mandell, J.B. Mandell. Breast and Ovarian Cancer Risks Due to Inherited Mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef]
- GenomeAsia 100K Consortium. The GenomeAsia 100K Project Enables Genetic Discoveries across Asia. Nature 2019, 576, 106–111. [Google Scholar] [CrossRef]
- Helgason, A.; Pálsson, S.; Thorleifsson, G.; Grant, S.F.; Emilsson, V.; Gunnarsdottir, S.; Adeyemo, A.; Chen, Y.; Chen, G.; Reynisdottir, I.; et al. Refining the Impact of TCF7L2 Gene Variants on Type 2 Diabetes and Adaptive Evolution. Nat. Genet. 2007, 39, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.; Vollger, M.R.; Dang, V.; Porubsky, D.; Baker, C.; Cantsilieris, S.; Hoekzema, K.; Lewis, A.P.; Munson, K.M.; Sorensen, M.; et al. Adaptive Archaic Introgression of Copy Number Variants and the Discovery of Previously Unknown Human Genes. Science 2019, 366. [Google Scholar] [CrossRef] [PubMed]
- Jackson, L.; Kuhlman, C.; Jackson, F.; Fox, K. Including Vulnerable Populations in the Assessment of Data From Vulnerable Populations. Front. Big Data 2019, 2. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fox, K.; Rallapalli, K.L.; Komor, A.C. Rewriting Human History and Empowering Indigenous Communities with Genome Editing Tools. Genes 2020, 11, 88. https://doi.org/10.3390/genes11010088
Fox K, Rallapalli KL, Komor AC. Rewriting Human History and Empowering Indigenous Communities with Genome Editing Tools. Genes. 2020; 11(1):88. https://doi.org/10.3390/genes11010088
Chicago/Turabian StyleFox, Keolu, Kartik Lakshmi Rallapalli, and Alexis C. Komor. 2020. "Rewriting Human History and Empowering Indigenous Communities with Genome Editing Tools" Genes 11, no. 1: 88. https://doi.org/10.3390/genes11010088
APA StyleFox, K., Rallapalli, K. L., & Komor, A. C. (2020). Rewriting Human History and Empowering Indigenous Communities with Genome Editing Tools. Genes, 11(1), 88. https://doi.org/10.3390/genes11010088