Off Earth Identification of Bacterial Populations Using 16S rDNA Nanopore Sequencing

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
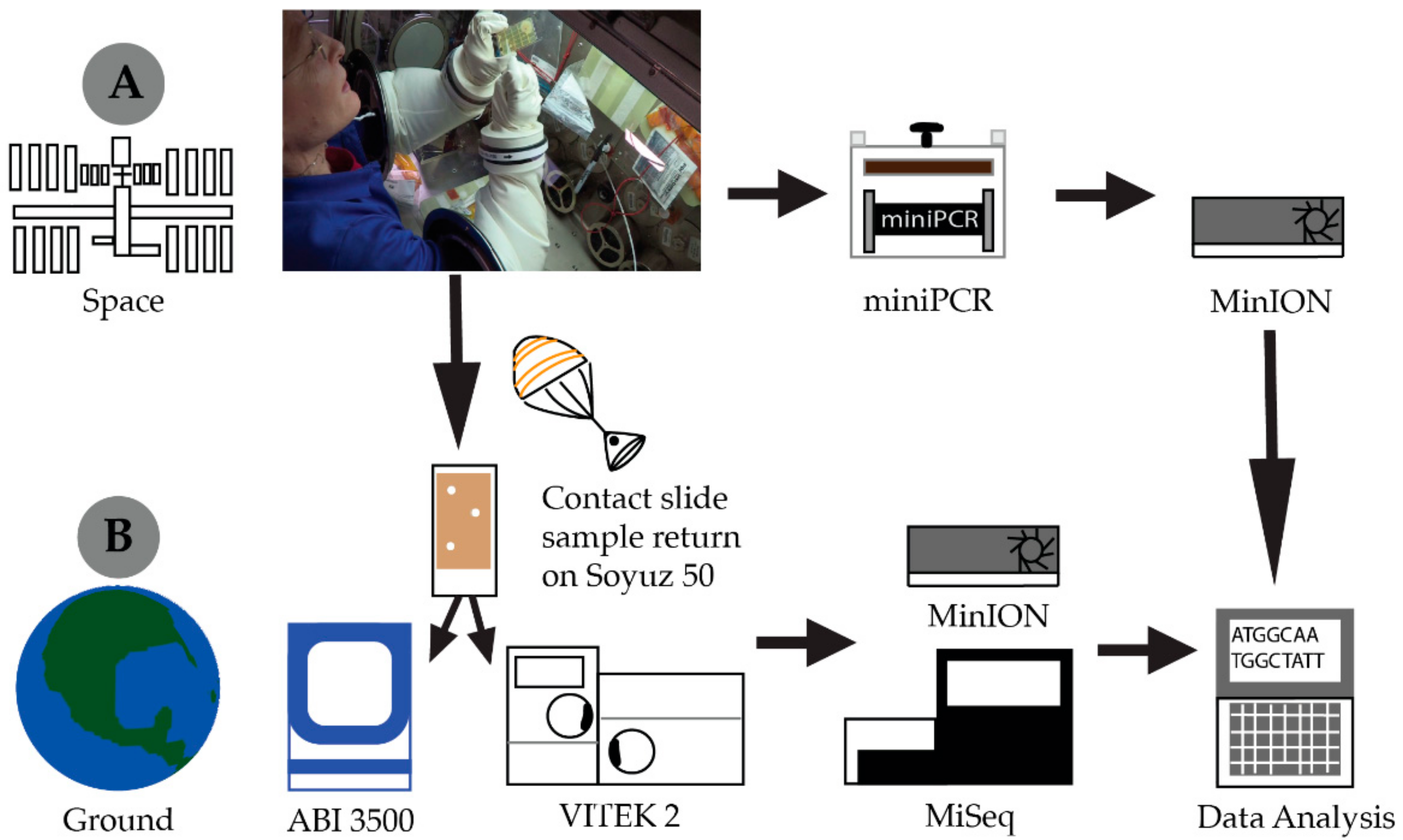
2.1. Spaceflight Hardware
2.2. Library Preparation and Sequencing (Ground and ISS)
2.2.1. 16S Amplification of DNA Standard
2.2.2. Microbial Collection and Culture
2.2.3. Cell Lysis and 16S Amplification
2.2.4. Library Preparation and Nanopore Sequencing
2.2.5. Ground Processing and Identification of the SSK Contact Slide
2.2.6. Ground Controls
2.3. Data Analysis
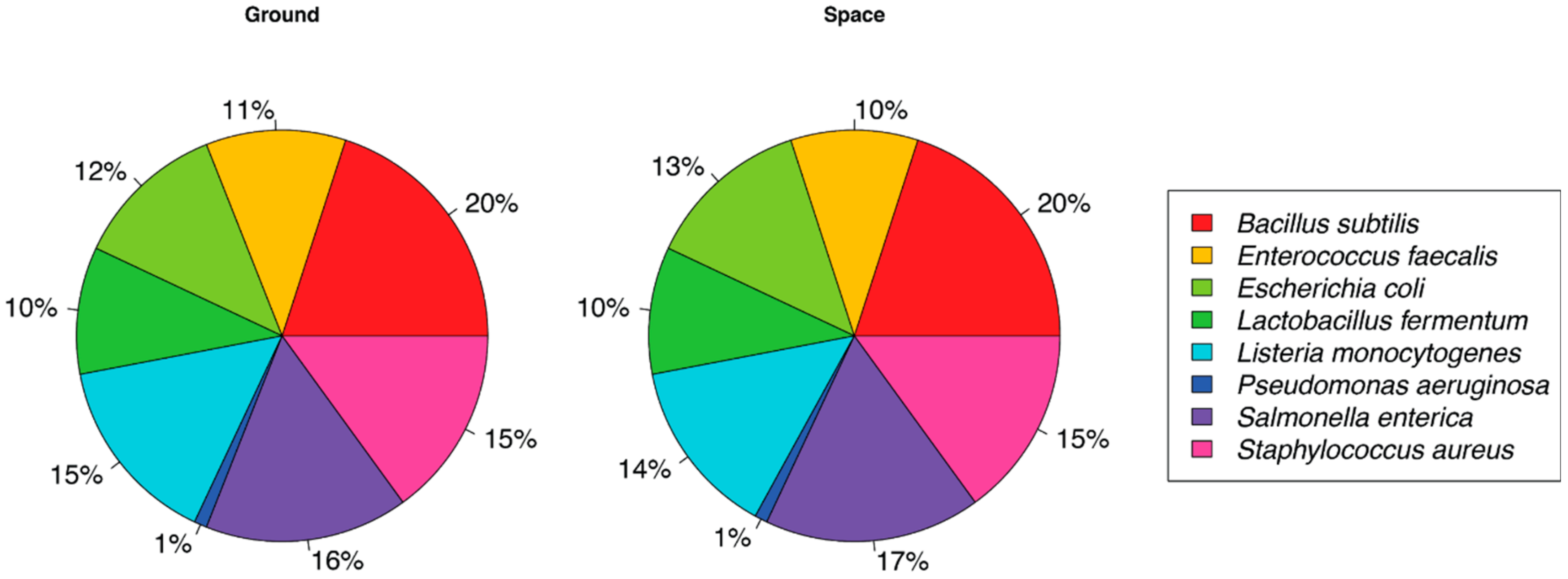
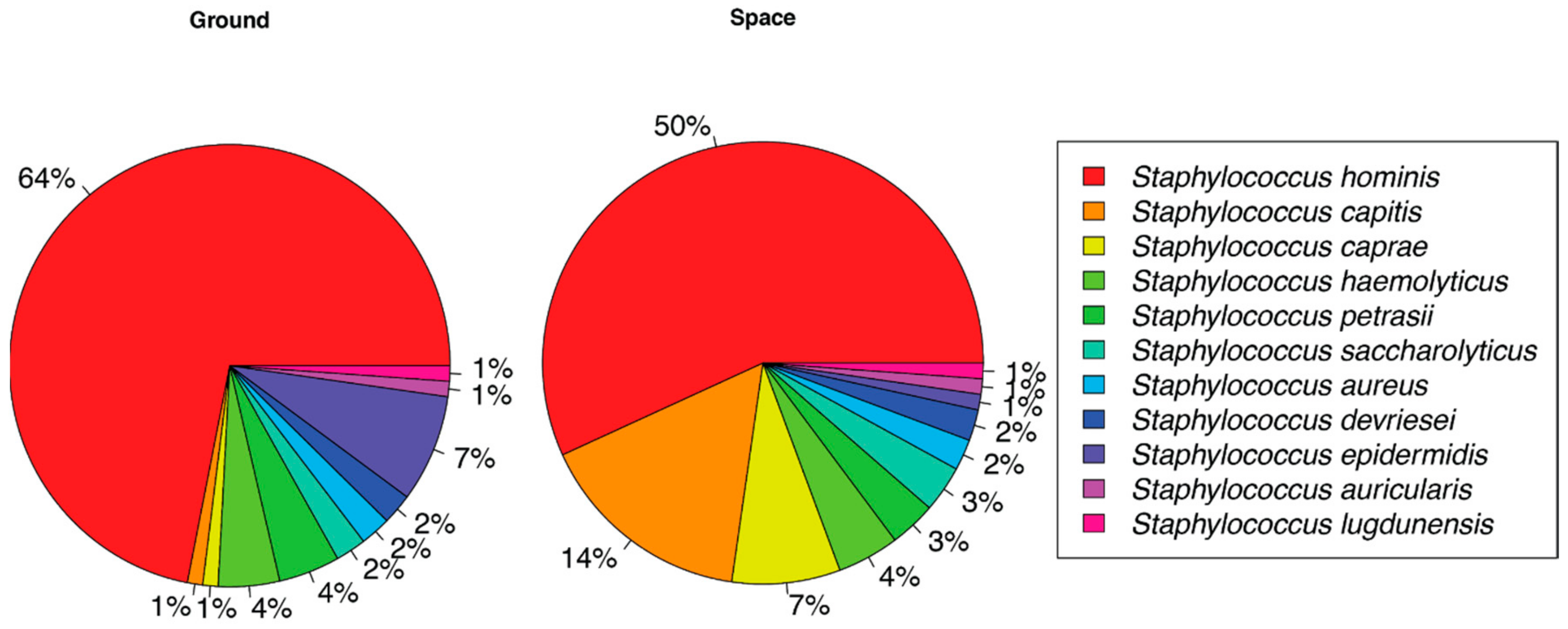
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Castro, A.V.; Thrasher, A.N.; Healy, M.; Ott, C.M.; Pierson, D.L. Microbial characterization during the early habitation of the International Space Station. Microb. Ecol. 2004, 47, 119–126. [Google Scholar] [CrossRef]
- Faria, N.R.; Sabino, E.C.; Nunes, M.R.; Alcantara, L.C.; Loman, N.J.; Pybus, O.G. Mobile real-time surveillance of Zika virus in Brazil. Genome. Med. 2016, 8, 97. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.; Debbonaire, A.R.; Nicholls, S.M.; Rassner, S.M.E.; Sattler, B.; Cook, J.M.; Davy, T.; Soares, A.; Mur, L.A.J.; Hodson, A.J. In-field metagenome and 16S rRNA gene amplicon nanopore sequencing robustly characterize glacier microbiota. bioRxiv 2019. [CrossRef]
- Johnson, S.S.; Zaikova, E.; Goerlitz, D.S.; Bai, Y.; Tighe, S.W. Real-Time DNA Sequencing in the Antarctic Dry Valleys Using the Oxford Nanopore Sequencer. J. Biomol. Tech. 2017, 28, 2–7. [Google Scholar] [CrossRef]
- Gowers, G.F.; Vince, O.; Charles, J.H.; Klarenberg, I.; Ellis, T.; Edward, A. Entirely Off-Grid and Solar-Powered DNA Sequencing of Microbial Communities during an Ice Cap Traverse Expedition. Genes (Basel) 2019, 10, 902. [Google Scholar] [CrossRef]
- Menegon, M.; Cantaloni, C.; Rodriguez-Prieto, A.; Centomo, C.; Abdelfattah, A.; Rossato, M.; Bernardi, M.; Xumerle, L.; Loader, S.; Delledonne, M. On site DNA barcoding by nanopore sequencing. PLoS ONE 2017, 12, e0184741. [Google Scholar] [CrossRef]
- Pomerantz, A.; Penafiel, N.; Arteaga, A.; Bustamante, L.; Pichardo, F.; Coloma, L.A.; Barrio-Amoros, C.L.; Salazar-Valenzuela, D.; Prost, S. Real-time DNA barcoding in a rainforest using nanopore sequencing: Opportunities for rapid biodiversity assessments and local capacity building. Gigascience 2018, 7, 33. [Google Scholar] [CrossRef]
- Boykin, L.M.; Sseruwagi, P.; Alicai, T.; Ateka, E.; Mohammed, I.U.; Stanton, J.L.; Kayuki, C.; Mark, D.; Fute, T.; Erasto, J.; et al. Tree Lab: Portable genomics for Early Detection of Plant Viruses and Pests in Sub-Saharan Africa. Genes (Basel) 2019, 10, 632. [Google Scholar] [CrossRef]
- Istace, B.; Friedrich, A.; d’Agata, L.; Faye, S.; Payen, E.; Beluche, O.; Caradec, C.; Davidas, S.; Cruaud, C.; Liti, G.; et al. de novo assembly and population genomic survey of natural yeast isolates with the Oxford Nanopore MinION sequencer. Gigascience 2017, 6, 1–13. [Google Scholar] [CrossRef]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Bolisetty, M.T.; Rajadinakaran, G.; Graveley, B.R. Determining exon connectivity in complex mRNAs by nanopore sequencing. Genome Biol. 2015, 16, 204. [Google Scholar] [CrossRef] [PubMed]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Jenjaroenpun, P.; Wongsurawat, T.; Pereira, R.; Patumcharoenpol, P.; Ussery, D.W.; Nielsen, J.; Nookaew, I. Complete genomic and transcriptional landscape analysis using third-generation sequencing: A case study of Saccharomyces cerevisiae CEN.PK113-7D. Nucleic Acids Res. 2018, 46, e38. [Google Scholar] [CrossRef] [PubMed]
- Sutton, M.A.; Burton, A.S.; Zaikova, E.; Sutton, R.E.; Brinckerhoff, W.B.; Bevilacqua, J.G.; Weng, M.M.; Mumma, M.J.; Johnson, S.S. Radiation Tolerance of Nanopore Sequencing Technology for Life Detection on Mars and Europa. Sci. Rep. 2019, 9, 5370. [Google Scholar] [CrossRef]
- Castro-Wallace, S.L.; Chiu, C.Y.; John, K.K.; Stahl, S.E.; Rubins, K.H.; McIntyre, A.B.R.; Dworkin, J.P.; Lupisella, M.L.; Smith, D.J.; Botkin, D.J.; et al. Nanopore DNA Sequencing and Genome Assembly on the International Space Station. Sci. Rep. 2017, 7, 18022. [Google Scholar] [CrossRef]
- Rizzardi, L.F.; Kunz, H.; Rubins, K.; Chouker, A.; Quiriarte, H.; Sams, C.; Crucian, B.E.; Feinberg, A.P. Evaluation of techniques for performing cellular isolation and preservation during microgravity conditions. Npj Microgravity 2016, 2, 16025. [Google Scholar] [CrossRef]
- Braman, K.M. Inter-Module Ventilation Changes to the International Space Station Vehicle to Support the Bigelow Expandable Activity Module. In 48th International Conference on Environmental Systems; ICES: Albuquerque, NM, USA, 2018. [Google Scholar]
- Roark, W.J.; Baugher, C.R.; Cockrell, D.W.; Gagliano, L.S. Microgravity Science Glovebox in 37th AIAA Aerospace Sciences Meeting and Exhibit; American Institute of Aeronautics and Astronautics: Reno, NV, USA, 1999. [Google Scholar]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquatic Micro. Ecology 2015, 75, 129–137. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4516–4522. [Google Scholar] [CrossRef]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.; Hyde, E.R.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.A.; et al. Improved Bacterial 16S rRNA Gene (V4 and V4-5) and Fungal Internal Transcribed Spacer Marker Gene Primers for Microbial Community Surveys. Msystems 2016, 1, e9–e15. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Fiddes, I.T.; Miga, K.H.; Olsen, H.E.; Paten, B.; Akeson, M. Improved data analysis for the MinION nanopore sequencer. Nat. Methods 2015, 12, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome. Biol. 2014, 15, R46. [Google Scholar] [CrossRef]
- Crucian, B.E.; Stowe, R.P.; Pierson, D.L.; Sams, C.F. Immune system dysregulation following short- vs long-duration spaceflight. Aviat. Space. Environ. Med. 2008, 79, 835–843. [Google Scholar] [CrossRef]
- Smith, S.M.; Heer, M.A.; Shackelford, L.C.; Sibonga, J.D.; Ploutz-Snyder, L.; Zwart, S.R. Benefits for bone from resistance exercise and nutrition in long-duration spaceflight: Evidence from biochemistry and densitometry. J. Bone. Miner. Res. 2012, 27, 1896–1906. [Google Scholar] [CrossRef]
- Wilson, J.W.; Ott, C.M.; Bentrup, K.H.z.; Ramamurthy, R.; Quick, L.; Porwollik, S.; Cheng, P.; McClelland, M.; Tsaprailis, G.; Radabaugh, T.; et al. Space flight alters bacterial gene expression and virulence and reveals a role for global regulator Hfq. Proc. Natl. Acad. Sci. USA 2007, 104, 16299–16304. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Colony | Detection Method | Sample ID | %ID |
|---|---|---|---|
| 1 | Biochemical | Staphylococcus hominis hominis | 97.0 |
| 2 | Biochemical | Staphylococcus hominis hominis | 97.0 |
| 3 | Biochemical | Staphylococcus capitis | 94.0 |
| 1 | Sanger Sequencing | Staphylococcus hominis hominis (ATCC = 27,844) | 99.9 |
| 2 | Sanger Sequencing | Staphylococcus hominis hominis (ATCC = 27,844) | 100.0 |
| 3 | Sanger Sequencing | Staphylococcus capitis capitis (ATCC = 27,840) | 99.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burton, A.S.; Stahl, S.E.; John, K.K.; Jain, M.; Juul, S.; Turner, D.J.; Harrington, E.D.; Stoddart, D.; Paten, B.; Akeson, M.; et al. Off Earth Identification of Bacterial Populations Using 16S rDNA Nanopore Sequencing. Genes 2020, 11, 76. https://doi.org/10.3390/genes11010076
Burton AS, Stahl SE, John KK, Jain M, Juul S, Turner DJ, Harrington ED, Stoddart D, Paten B, Akeson M, et al. Off Earth Identification of Bacterial Populations Using 16S rDNA Nanopore Sequencing. Genes. 2020; 11(1):76. https://doi.org/10.3390/genes11010076
Chicago/Turabian StyleBurton, Aaron S., Sarah E. Stahl, Kristen K. John, Miten Jain, Sissel Juul, Daniel J. Turner, Eoghan D. Harrington, David Stoddart, Benedict Paten, Mark Akeson, and et al. 2020. "Off Earth Identification of Bacterial Populations Using 16S rDNA Nanopore Sequencing" Genes 11, no. 1: 76. https://doi.org/10.3390/genes11010076
APA StyleBurton, A. S., Stahl, S. E., John, K. K., Jain, M., Juul, S., Turner, D. J., Harrington, E. D., Stoddart, D., Paten, B., Akeson, M., & Castro-Wallace, S. L. (2020). Off Earth Identification of Bacterial Populations Using 16S rDNA Nanopore Sequencing. Genes, 11(1), 76. https://doi.org/10.3390/genes11010076