Large-Scale Profiling of RBP-circRNA Interactions from Public CLIP-Seq Datasets


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Downloading CLIP-Seq Datasets
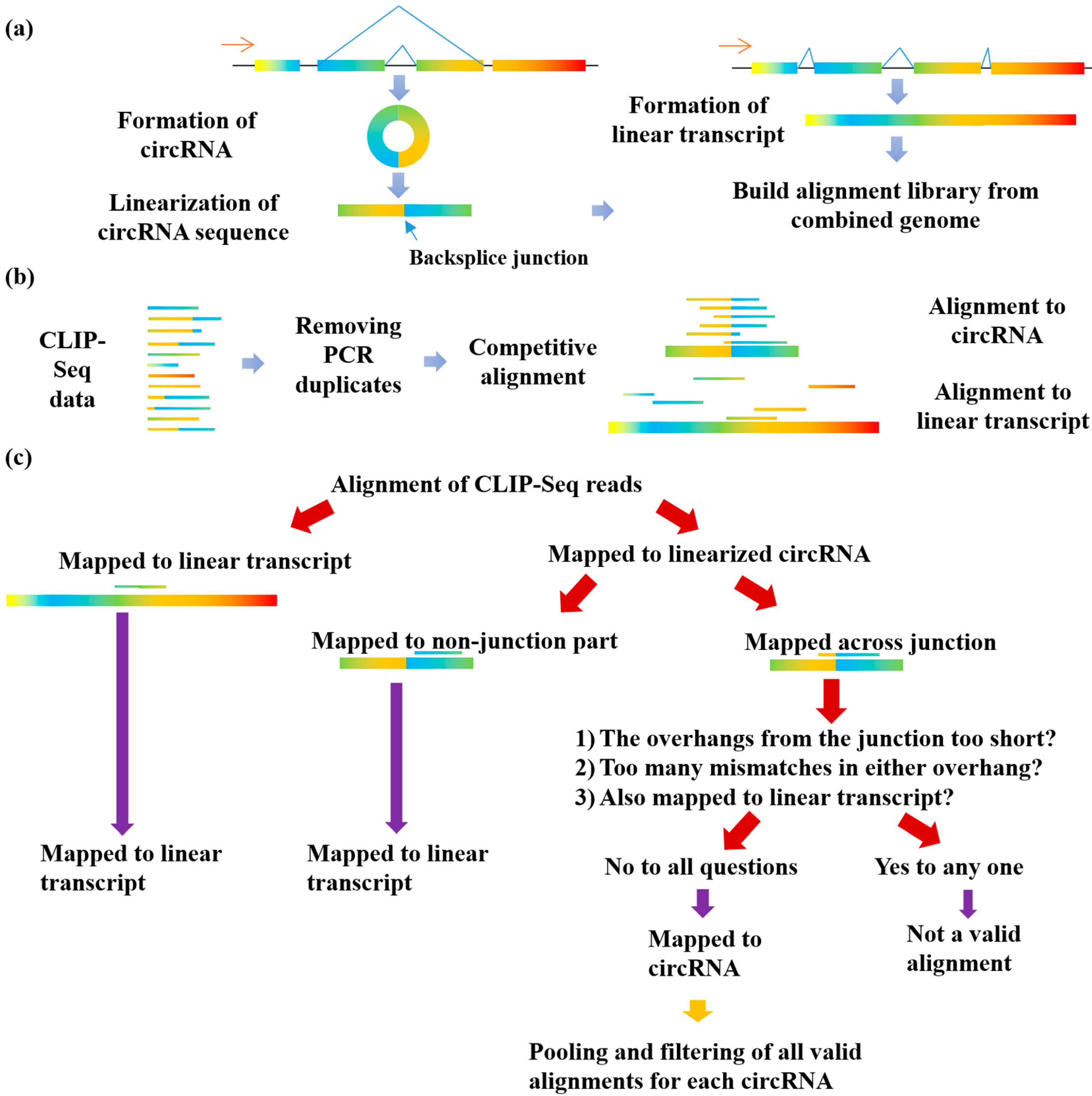
2.2. Linearization of circRNA Library
2.3. Competitive Alignment of CLIP-Seq Reads
2.4. Filtering for CLIP-Seq Reads Supporting RBP-circRNA Interactions
2.5. Software Implementation
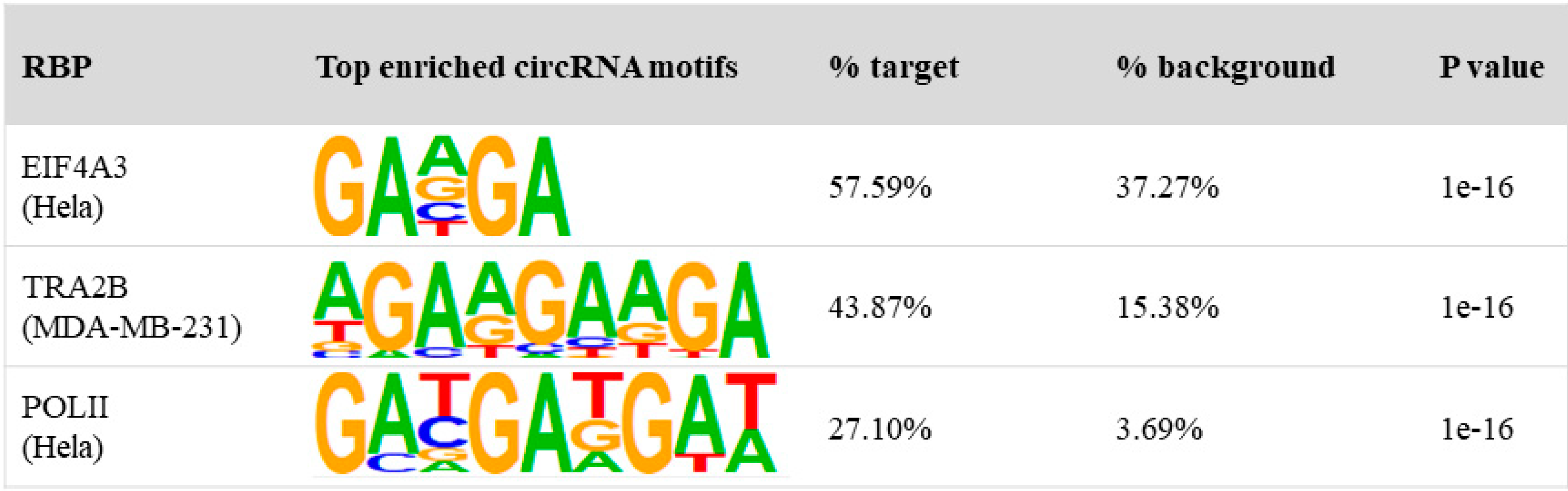
2.6. Motif Search
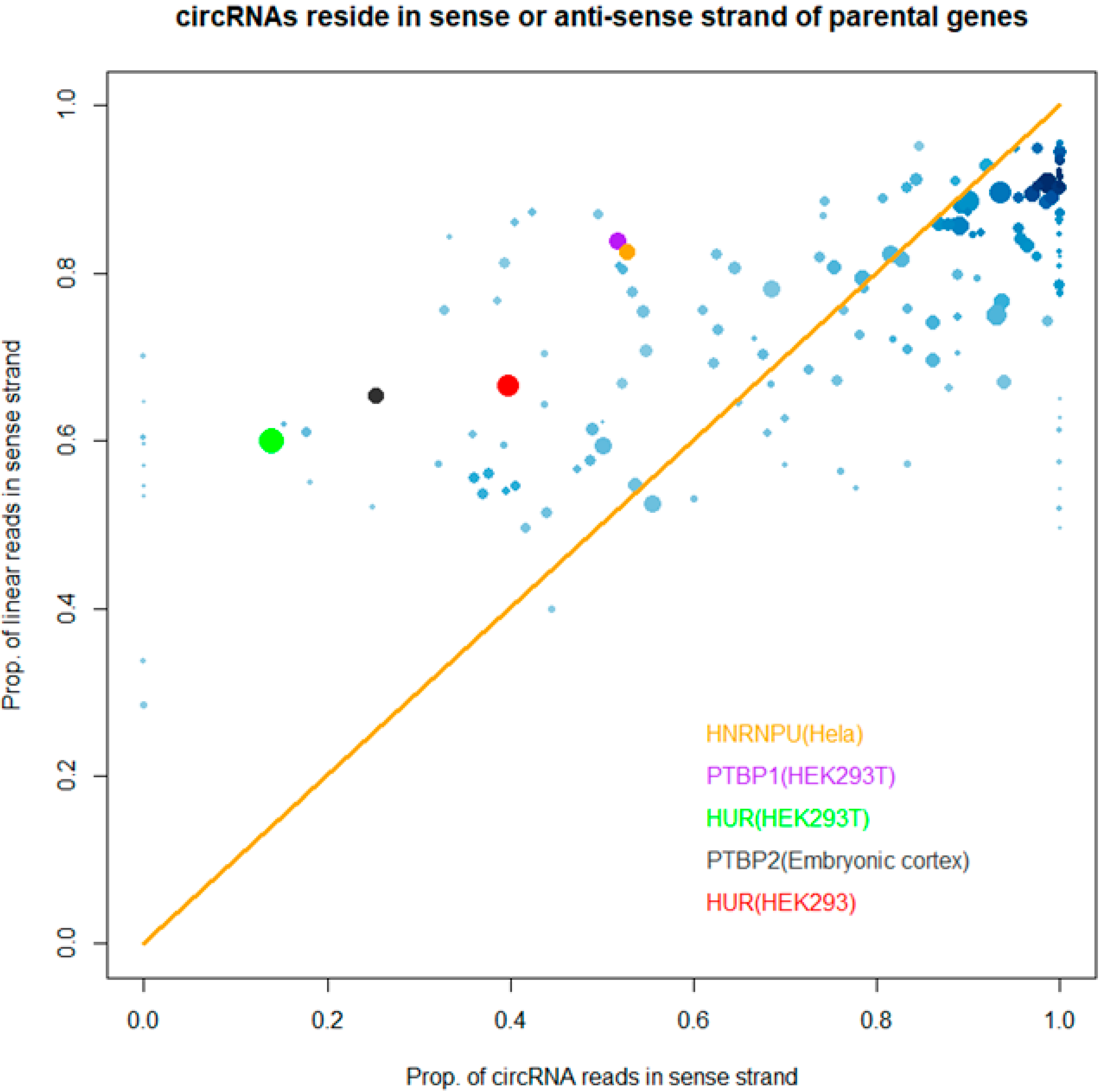
2.7. “Strand Bias” of RBP Binding to circRNAs
2.8. Hierarchical Clustering of RBPs
2.9. Gene Ontology Enrichment Analysis
3. Results
3.1. The Implementation of the Clirc Software
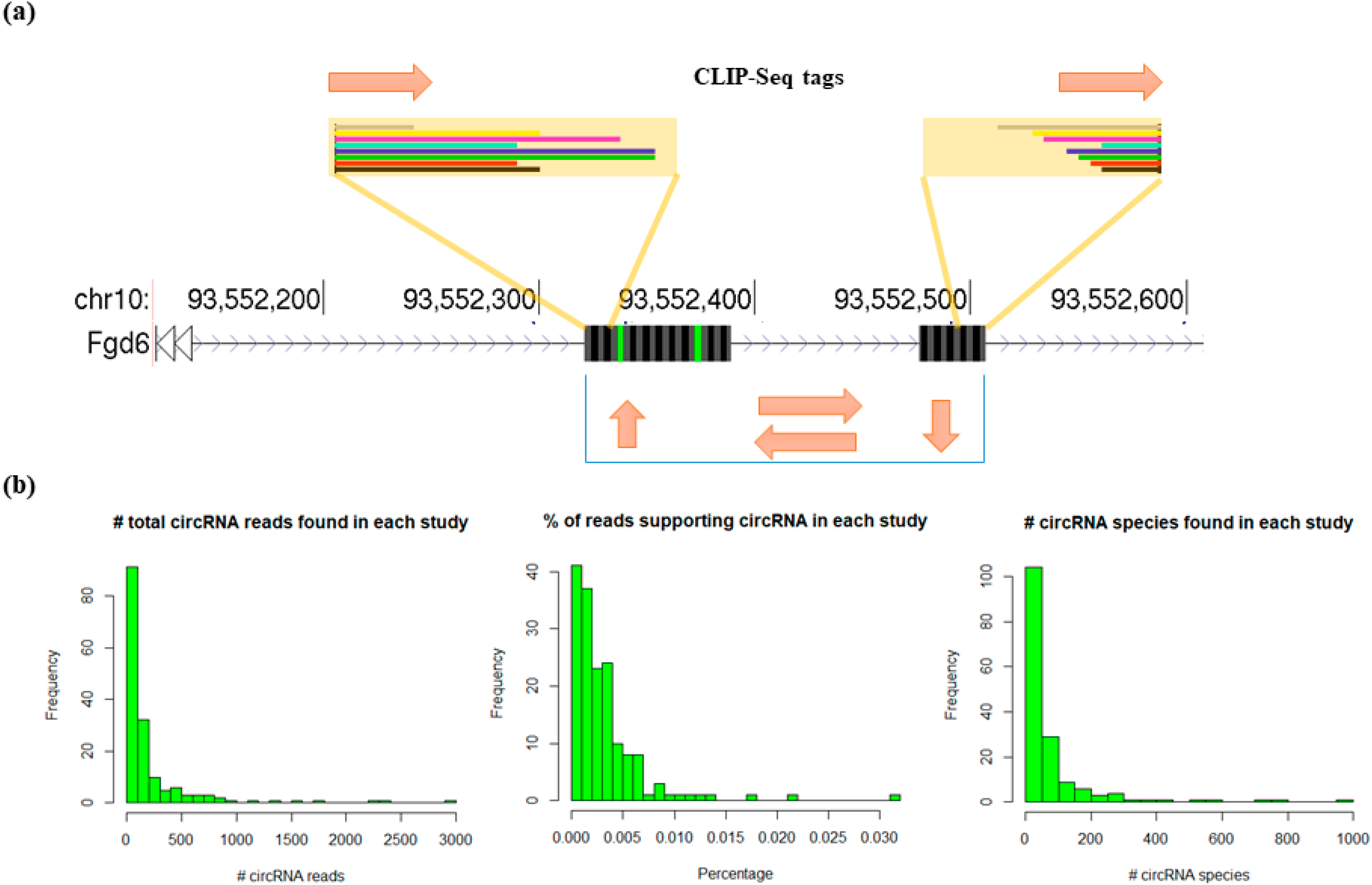
3.2. Profiling circRNAs Bound by RBPs Using Clirc
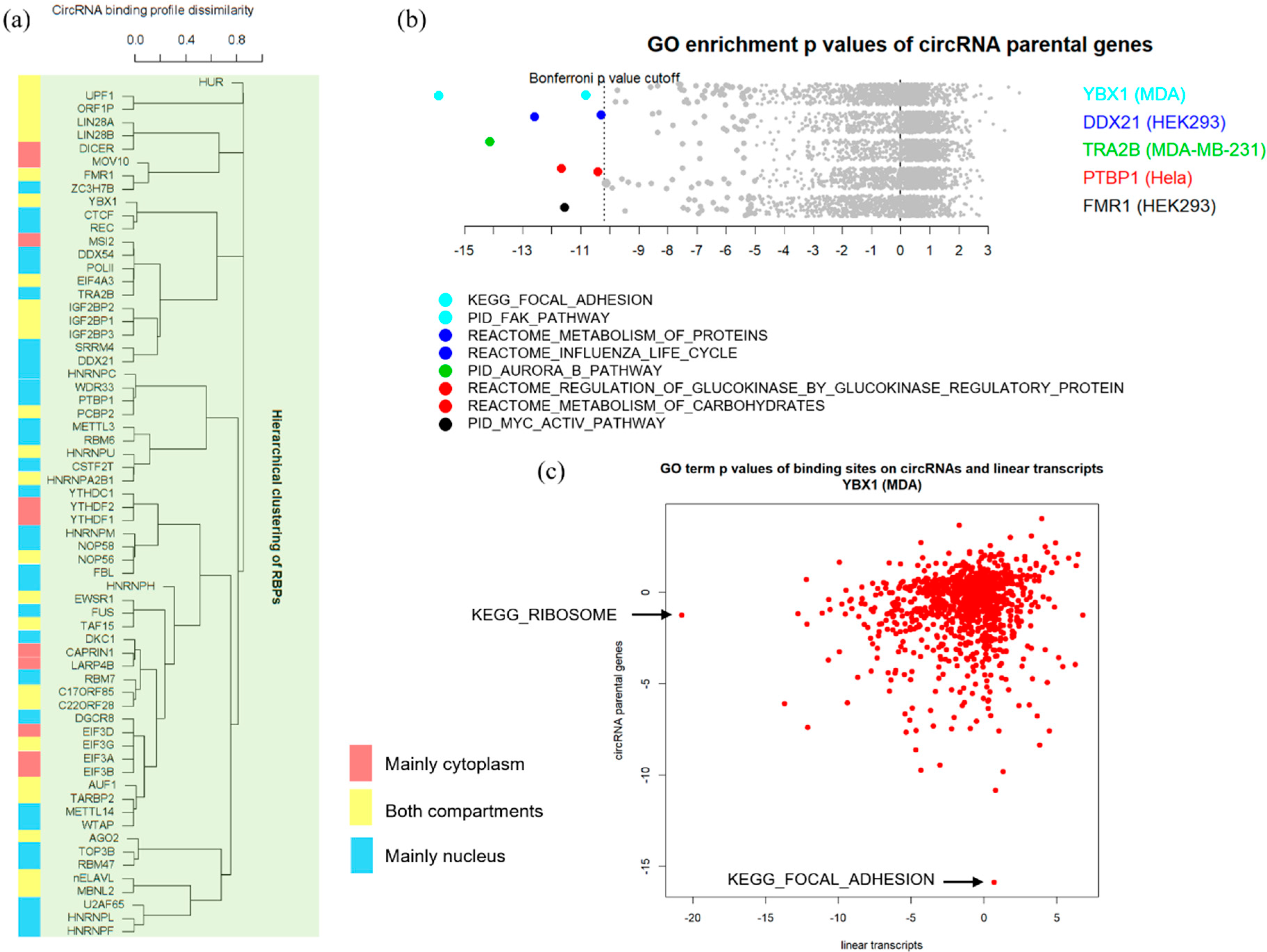
3.3. Binding Properties of RBPs on circRNAs
3.4. Functional Implications of circRNA-RBP Interactions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Loewer, A. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Starke, S.; Jost, I.; Rossbach, O.; Schneider, T.; Schreiner, S.; Hung, L.H.; Bindereif, A. Exon circularization requires canonical splice signals. Cell Rep. 2015, 10, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Z. Efficient backsplicing produces translatable circular mRNAs. RNA 2015, 21, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Agarwal, V.; Guo, H.; Bartel, D.P. Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 2014, 15, 409. [Google Scholar] [CrossRef] [PubMed]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. CircRNA biogenesis competes with pre-mRNA splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Conn, S.J.; Pillman, K.A.; Toubia, J.; Conn, V.M.; Salmanidis, M.; Phillips, C.A.; Roslan, S.; Schreiber, A.W.; Gregory, P.A.; Goodall, G.J. The RNA Binding Protein Quaking Regulates Formation of circRNAs. Cell 2015, 160, 1125–1134. [Google Scholar] [CrossRef]
- You, X.; Vlatkovic, I.; Babic, A.; Will, T.; Epstein, I.; Tushev, G.; Akbalik, G.; Wang, M.; Glock, C.; Quedenau, C.; et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat. Neurosci. 2015, 18, 603–610. [Google Scholar] [CrossRef]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256–264. [Google Scholar] [CrossRef]
- Bachmayr-Heyda, A.; Reiner, A.T.; Auer, K.; Sukhbaatar, N.; Aust, S.; Bachleitner-Hofmann, T.; Mesteri, I.; Grunt, T.W.; Zeillinger, R.; Pils, D. Correlation of circular RNA abundance with proliferation—Exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci. Rep. 2015, 5, 8057. [Google Scholar] [CrossRef]
- Valdmanis, P.N.; Kay, M.A. The expanding repertoire of circular RNAs. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 1112–1114. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Jensen, K.B.; Ruggiu, M.; Mele, A.; Ule, A.; Darnell, R.B. CLIP identifies Nova-regulated RNA networks in the brain. Science 2003, 302, 1212–1215. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Xiao, G.; Chu, Y.; Zhang, M.Q.; Corey, D.R.; Xie, Y. Design and bioinformatics analysis of genome-wide CLIP experiments. Nucleic Acids Res. 2015, 43, 5263–5274. [Google Scholar] [CrossRef] [PubMed]
- Licatalosi, D.D.; Mele, A.; Fak, J.J.; Ule, J.; Kayikci, M.; Chi, S.W.; Clark, T.A.; Schweitzer, A.C.; Blume, J.E.; Wang, X.; et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 2008, 456, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M.; Jungkamp, A.C.; Munschauer, M.; et al. PAR-CliP—A method to identify transcriptome-wide the binding sites of RNA binding proteins. J. Vis. Exp. 2010, 41, e2034. [Google Scholar] [CrossRef]
- Konig, J.; Zarnack, K.; Rot, G.; Curk, T.; Kayikci, M.; Zupan, B.; Turner, D.J.; Luscombe, N.M.; Ule, J. ICLIP—Transcriptome-wide mapping of protein-RNA interactions with individual nucleotide resolution. J. Vis. Exp. 2011, 50, e2638. [Google Scholar] [CrossRef]
- Van Nostrand, E.L.; Pratt, G.A.; Shishkin, A.A.; Gelboin-Burkhart, C.; Fang, M.Y.; Sundararaman, B.; Stanton, R. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 2016, 13, 508. [Google Scholar] [CrossRef]
- Kargapolova, Y.; Levin, M.; Lackner, K.; Danckwardt, S. SCLIP—An integrated platform to study RNA–Protein interactomes in biomedical research: Identification of CSTF2tau in alternative processing of small nuclear RNAs. Nucleic Acids Res. 2017, 45, 6074–6086. [Google Scholar] [CrossRef]
- Zarnegar, B.J.; Flynn, R.A.; Shen, Y.; Do, B.T.; Chang, H.Y.; Khavari, P.A. IrCLIP platform for efficient characterization of protein–RNA interactions. Nat. Methods 2016, 13, 489. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, J.; Zhao, F. Circular RNA identification based on multiple seed matching. Brief. Bioinform. 2017, 19, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Glazar, P.; Papavasileiou, P.; Rajewsky, N. CircBase: A database for circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Mouse, E.C.; Stamatoyannopoulos, J.A.; Snyder, M.; Hardison, R.; Ren, B.; Gingeras, T.; Gilbert, D.M.; Groudine, M.; Bender, M.; Kaul, R.; et al. An encyclopedia of mouse DNA elements (Mouse ENCODE). Genome Biol. 2012, 13, 418. [Google Scholar] [CrossRef] [PubMed]
- Mod, E.C.; Roy, S.; Ernst, J.; Kharchenko, P.V.; Kheradpour, P.; Negre, N.; Eaton, M.L.; Landolin, J.M.; Bristow, C.A.; Ma, L.; et al. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 2010, 330, 1787–1797. [Google Scholar]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef]
- Wu, T.D.; Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 2010, 26, 873–881. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Mesirov, J.P. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Westholm, J.O.; Miura, P.; Olson, S.; Shenker, S.; Joseph, B.; Sanfilippo, P.; Celniker, S.E.; Graveley, B.R.; Lai, E.C. Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep. 2014, 9, 1966–1980. [Google Scholar] [CrossRef]
- Rybak-Wolf, A.; Stottmeister, C.; Glazar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartok, O.; Ashwal-Fluss, R.; et al. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol. Cell 2015, 58, 870–885. [Google Scholar] [CrossRef] [PubMed]
- Kechris, K.; Yang, Y.H.; Yeh, R.F. Prediction of alternatively skipped exons and splicing enhancers from exon junction arrays. BMC Genom. 2008, 9, 551. [Google Scholar] [CrossRef] [PubMed]
- Caudevilla, C.; Codony, C.; Serra, D.; Plasencia, G.; Roman, R.; Graessmann, A.; Asins, G.; Bach-Elias, M.; Hegardt, F.G. Localization of an exonic splicing enhancer responsible for mammalian natural trans-splicing. Nucleic Acids Res. 2001, 29, 3108–3115. [Google Scholar] [CrossRef] [PubMed]
- Haremaki, T.; Weinstein, D.C. Eif4a3 is required for accurate splicing of the Xenopus laevis ryanodine receptor pre-mRNA. Dev. Biol. 2012, 372, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Dichmann, D.S.; Walentek, P.; Harland, R.M. The alternative splicing regulator Tra2b is required for somitogenesis and regulates splicing of an inhibitory Wnt11b isoform. Cell Rep. 2015, 10, 527–536. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef]
- Wang, Y.; Su, J.; Fu, D.; Wang, Y.; Chen, Y.; Chen, R.; Yue, D. The Role of YB1 in Renal Cell Carcinoma Cell Adhesion. Int. J. Med. Sci. 2018, 15, 1304. [Google Scholar] [CrossRef]
- Kang, Y.; Hu, W.; Ivan, C.; Dalton, H.J.; Miyake, T.; Pecot, C.V.; Rupaimoole, R. Role of focal adhesion kinase in regulating YB–1–mediated paclitaxel resistance in ovarian cancer. J. Natl. Cancer Inst. 2013, 105, 1485–1495. [Google Scholar] [CrossRef]
- Chen, G.; Liu, C.H.; Zhou, L.; Krug, R.M. Cellular DDX21 RNA helicase inhibits influenza A virus replication but is counteracted by the viral NS1 protein. Cell Host Microbe 2014, 15, 484–493. [Google Scholar] [CrossRef]
- Li, J.H.; Liu, S.; Zheng, L.L.; Wu, J.; Sun, W.J.; Wang, Z.L.; Yang, J.H. Discovery of protein–lncRNA interactions by integrating large-scale CLIP-Seq and RNA-Seq datasets. Front. Bioeng. Biotechnol. 2015, 2, 88. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://noncodingrnaexplorer.wordpress.com (accessed on 31 December 2019).





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Wang, T.; Xiao, G.; Xie, Y. Large-Scale Profiling of RBP-circRNA Interactions from Public CLIP-Seq Datasets. Genes 2020, 11, 54. https://doi.org/10.3390/genes11010054
Zhang M, Wang T, Xiao G, Xie Y. Large-Scale Profiling of RBP-circRNA Interactions from Public CLIP-Seq Datasets. Genes. 2020; 11(1):54. https://doi.org/10.3390/genes11010054
Chicago/Turabian StyleZhang, Minzhe, Tao Wang, Guanghua Xiao, and Yang Xie. 2020. "Large-Scale Profiling of RBP-circRNA Interactions from Public CLIP-Seq Datasets" Genes 11, no. 1: 54. https://doi.org/10.3390/genes11010054
APA StyleZhang, M., Wang, T., Xiao, G., & Xie, Y. (2020). Large-Scale Profiling of RBP-circRNA Interactions from Public CLIP-Seq Datasets. Genes, 11(1), 54. https://doi.org/10.3390/genes11010054