Developmental Transcriptomic Analysis of the Cave-Dwelling Crustacean, Asellus aquaticus

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
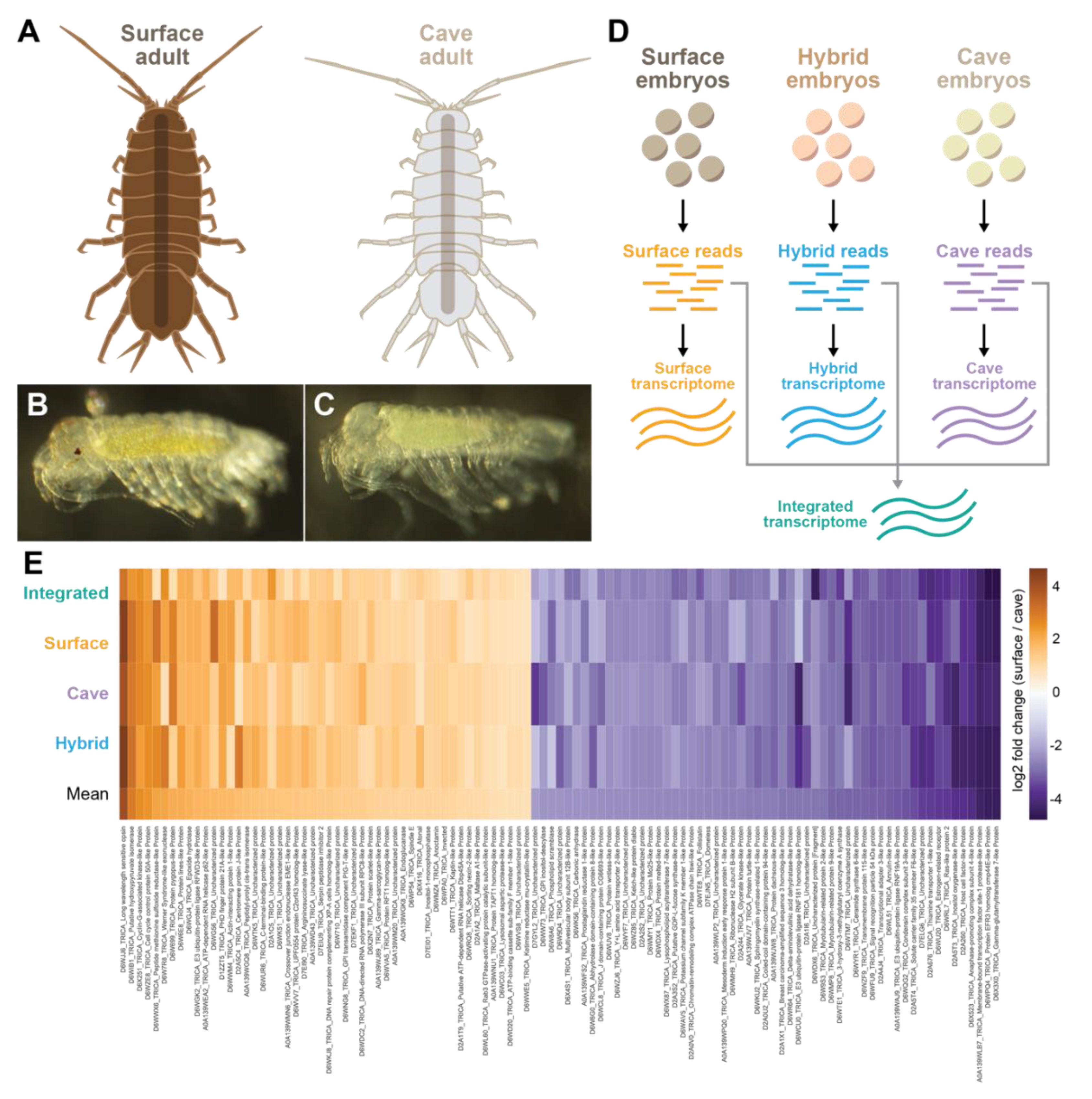
2.1. Animals
2.2. RNA Extraction, Library Preparation, and Sequencing
2.3. De Novo Transcriptome Assembly and Annotation
2.4. RNA-Sequencing and Expression Analyses
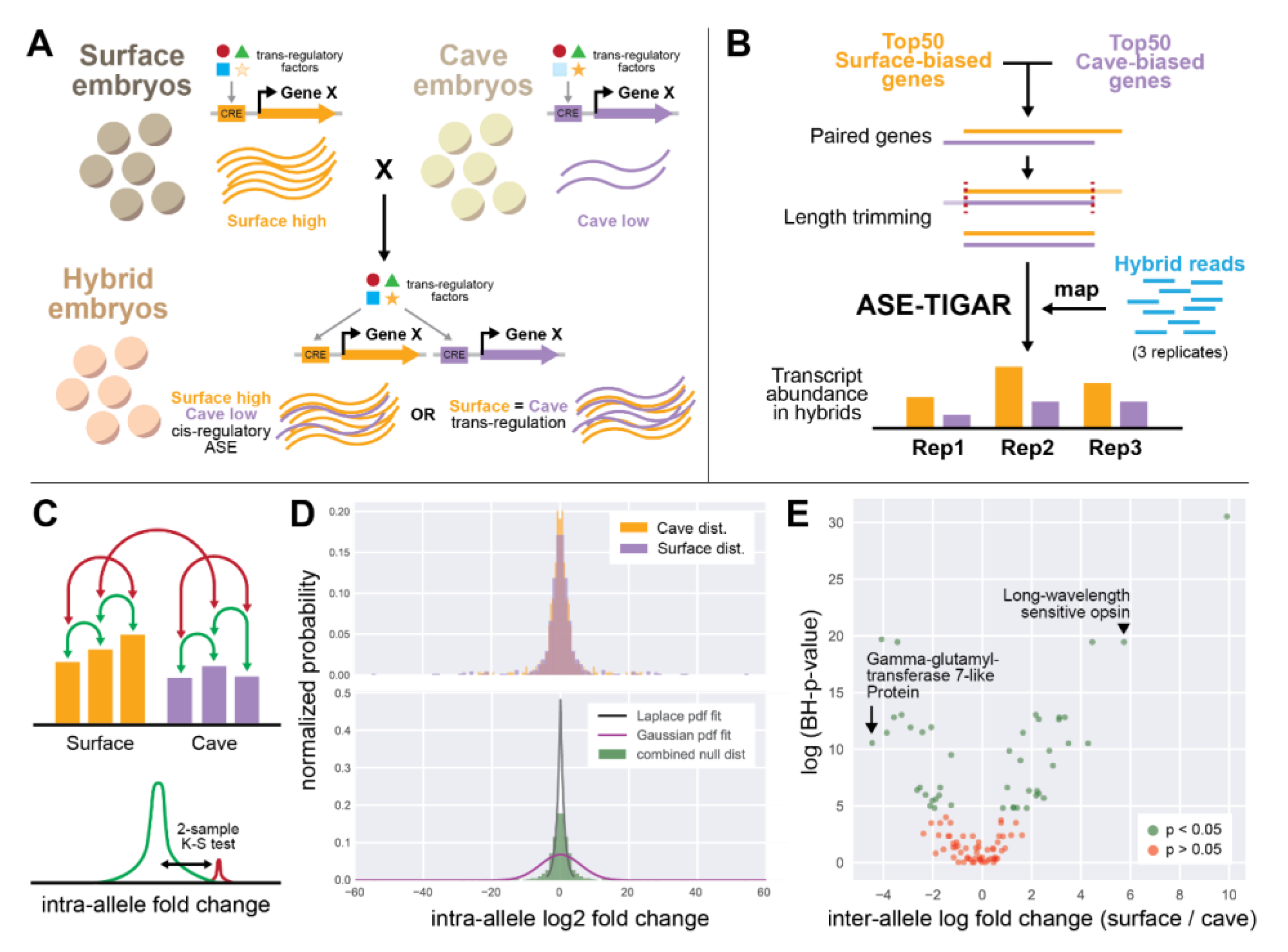
2.5. Allele-Specific Expression Using ASE-TIGAR
2.6. Data Deposition
3. Results
3.1. Characterization of Surface, Cave, and Hybrid Transcriptomes
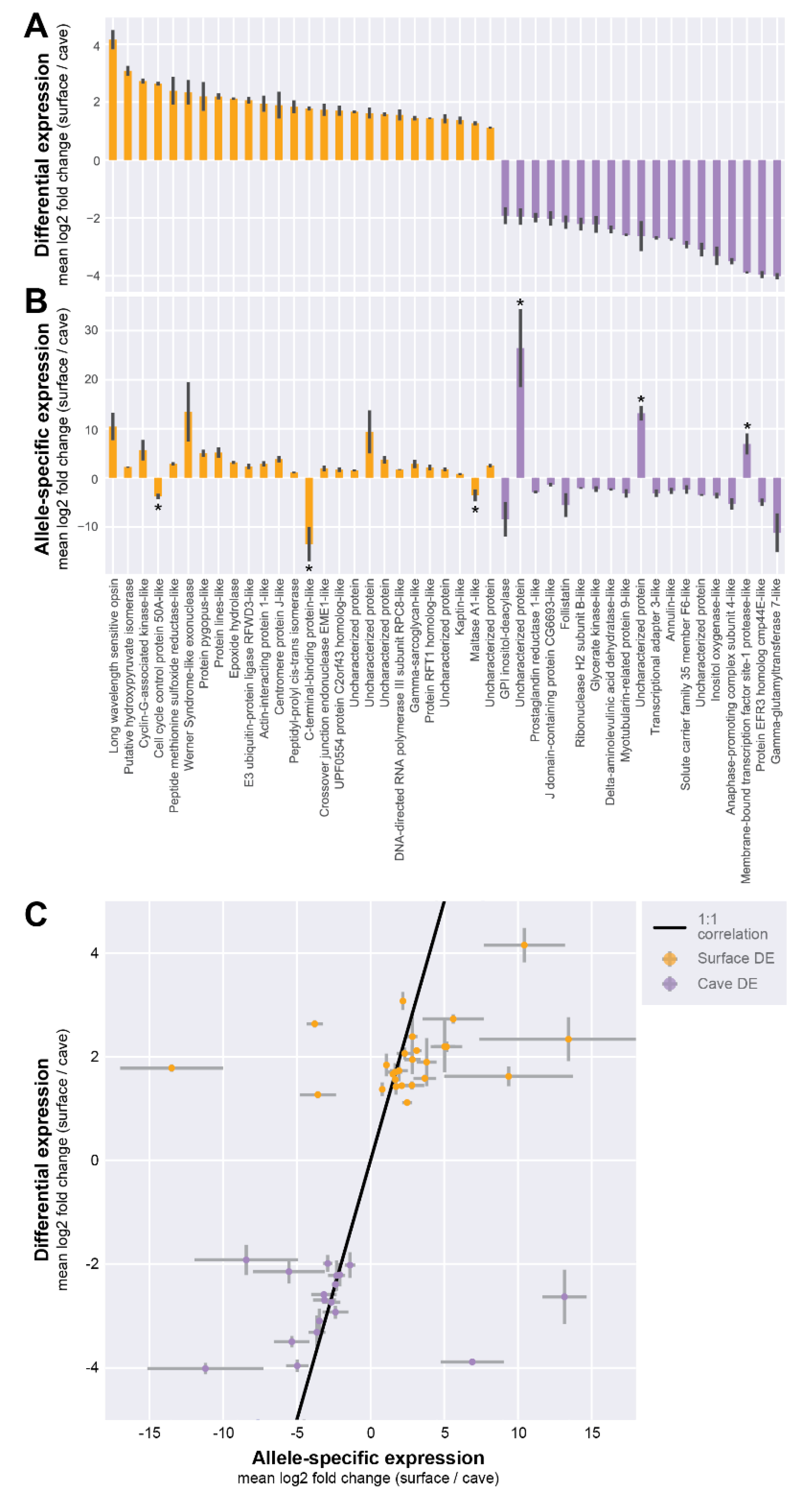
3.2. Differential RNA-Seq Analysis Between Cave and Surface Morphs
3.3. Allele-Specific Expression Analysis in Hybrid Individuals
4. Discussion
4.1. Candidate Genes
4.2. Involvement of Regulatory Mutation Versus Coding Mutation in Evolution of Cave Traits
4.3. Cis-Versus Trans-Regulation
4.4. Parent-of-Origin Effects Versus Cis-Regulation
4.5. Comparison to Adult Asellus Transcriptome
4.6. Comparison to Other Cave-Dwelling Animal Transcriptomes
4.7. Future Steps
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, J.; Chen, X.; Bai, J.; Fang, D.; Qiu, Y.; Jiang, W.; Yuan, H.; Bian, C.; Lu, J.; He, S.; et al. The Sinocyclocheilus Cavefish Genome Provides Insights into Cave Adaptation. BMC Biol. 2016, 14, 1–4. [Google Scholar] [CrossRef]
- McGaugh, S.E.; Gross, J.B.; Aken, B.; Blin, M.; Borowsky, R.; Chalopin, D.; Hinaux, H.; Jeffery, W.R.; Keene, A.; Ma, L.; et al. The Cavefish Genome Reveals Candidate Genes for Eye Loss. Nat. Commun. 2014, 5, 5307. [Google Scholar] [CrossRef]
- Carlini, D.B.; Satish, S.; Fong, D.W. Parallel Reduction in Expression, but no Loss of Functional Constraint, in Two Opsin Paralogs within Cave Populations of Gammarus Minus (Crustacea: Amphipoda). BMC Evol. Biol. 2013, 13, 89. [Google Scholar] [CrossRef]
- Meng, F.; Braasch, I.; Phillips, J.B.; Lin, X.; Titus, T.; Zhang, C.; Postlethwait, J.H. Evolution of the Eye Transcriptome Under Constant Darkness in Sinocyclocheilus Cavefish. Mol. Biol. Evol. 2013, 30, 1527–1543. [Google Scholar] [CrossRef]
- Passow, C.N.; Brown, A.P.; Arias-Rodriguez, L.; Yee, M.C.; Sockell, A.; Schartl, M.; Warren, W.C.; Bustamante, C.; Kelley, J.L.; Tobler, M. Complexities of Gene Expression Patterns in Natural Populations of an Extremophile Fish (Poecilia Mexicana, Poeciliidae). Mol. Ecol. 2017, 26, 4211–4225. [Google Scholar] [CrossRef]
- Stahl, B.A.; Gross, J.B.; Speiser, D.I.; Oakley, T.H.; Patel, N.H.; Gould, D.B.; Protas, M.E. A Transcriptomic Analysis of Cave, Surface, and Hybrid Isopod Crustaceans of the Species Asellus Aquaticus. PLoS ONE 2015, 10, e0140484. [Google Scholar] [CrossRef] [PubMed]
- Stern, D.B.; Crandall, K.A. Phototransduction Gene Expression and Evolution in Cave and Surface Crayfishes. Integr. Comp. Biol. 2018, 58, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Stern, D.B.; Crandall, K.A. The Evolution of Gene Expression Underlying Vision Loss in Cave Animals. Mol. Biol. Evol. 2018, 35, 2005–2014. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, W.R. Chapter 8. Evolution and Development in the Cavefish Astyanax. Curr. Top. Dev. Biol. 2009, 86, 191–221. [Google Scholar]
- Jeffery, W.R. Adaptive Evolution of Eye Degeneration in the Mexican Blind Cavefish. J. Hered. 2005, 96, 185–196. [Google Scholar] [CrossRef]
- Gross, J.B.; Furterer, A.; Carlson, B.M.; Stahl, B.A. An Integrated Transcriptome-Wide Analysis of Cave and Surface Dwelling Astyanax Mexicanus. PLoS ONE 2013, 8, e55659. [Google Scholar] [CrossRef] [PubMed]
- Hinaux, H.; Poulain, J.; Da Silva, C.; Noirot, C.; Jeffery, W.R.; Casane, D.; Retaux, S. De Novo Sequencing of Astyanax Mexicanus Surface Fish and Pachon Cavefish Transcriptomes Reveals Enrichment of Mutations in Cavefish Putative Eye Genes. PLoS ONE 2013, 8, e53553. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, H.; Wang, Y.; Adamski, K.; Rohner, N.; Kowalko, J.E. CRISPR Mutagenesis Confirms the Role of Oca2 in Melanin Pigmentation in Astyanax Mexicanus. Dev. Biol. 2018, 441, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Kowalko, J.E.; Ma, L.; Jeffery, W.R. Genome Editing in Astyanax Mexicanus using Transcription Activator-Like Effector Nucleases (TALENs). J. Vis. Exp. 2016, 112. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Jeffery, W.R.; Essner, J.J.; Kowalko, J.E. Genome Editing using TALENs in Blind Mexican Cavefish, Astyanax Mexicanus. PLoS ONE 2015, 10, e0119370. [Google Scholar] [CrossRef] [PubMed]
- Casane, D.; Retaux, S. Evolutionary Genetics of the Cavefish Astyanax Mexicanus. Adv. Genet. 2016, 95, 117–159. [Google Scholar]
- Gross, J.B.; Meyer, B.; Perkins, M. The Rise of Astyanax Cavefish. Dev. Dyn. 2015, 244, 1031–1038. [Google Scholar] [CrossRef]
- Protas, M.; Jeffery, W.R. Evolution and Development in Cave Animals: From Fish to Crustaceans. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 823–845. [Google Scholar] [CrossRef]
- Verovnik, R.; Sket, B.; Trontelj, P. Phylogeography of Subterranean and Surface Populations of Water Lice Asellus Aquaticus (Crustacea: Isopoda). Mol. Ecol. 2004, 13, 1519–1532. [Google Scholar] [CrossRef]
- Verovnik, R.; Sket, B.; Trontelj, P. The Colonization of Europe by the Freshwater Crustacean Asellus Aquaticus (Crustacea: Isopoda) Proceeded from Ancient Refugia and was Directed by Habitat Connectivity. Mol. Ecol. 2005, 14, 4355–4369. [Google Scholar] [CrossRef]
- Verovnik, R.; Sket, B.; Prevorcnik, S.; Trontelj, P. Random Amplified Polymorphic DNA Diversity among Surface and Subterranean Populations of Asellus Aquaticus (Crustacea: Isopoda). Genetica 2003, 119, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Turk, S.; Sket, B.; Sarbu, S. Comparison between some Epigean and Hypogean Populations of Asellus Aquaticus (Crustacea: Isopoda: Asellidae). Hydrobiologia 1996, 337, 161–170. [Google Scholar] [CrossRef]
- Turk-Prevorčnik, S.; Blejec, A. Asellus Aquaticus Infernus, New Subspecies (Isopoda: Asellota: Asellidae), from Romanian Hypogean Waters. J. Crustacean Biol. 1998, 18, 763–773. [Google Scholar] [CrossRef]
- Perez-Moreno, J.L.; Balazs, G.; Wilkins, B.; Herczeg, G.; Bracken-Grissom, H.D. The Role of Isolation on Contrasting Phylogeographic Patterns in Two Cave Crustaceans. BMC Evol. Biol. 2017, 17, 247–249. [Google Scholar] [CrossRef]
- Re, C.; Fiser, Z.; Perez, J.; Tacdol, A.; Trontelj, P.; Protas, M.E. Common Genetic Basis of Eye and Pigment Loss in Two Distinct Cave Populations of the Isopod Crustacean Asellus Aquaticus. Integr. Comp. Biol. 2018, 58, 421–430. [Google Scholar] [CrossRef]
- Protas, M.E.; Trontelj, P.; Patel, N.H. Genetic Basis of Eye and Pigment Loss in the Cave Crustacean, Asellus Aquaticus. Proc. Natl. Acad. Sci. USA 2011, 108, 5702–5707. [Google Scholar] [CrossRef]
- Mojaddidi, H.; Fernandez, F.E.; Erickson, P.A.; Protas, M.E. Embryonic Origin and Genetic Basis of Cave Associated Phenotypes in the Isopod Crustacean Asellus Aquaticus. Sci. Rep. 2018, 8, 16589. [Google Scholar] [CrossRef]
- Perez-Moreno, J.L.; Balazs, G.; Bracken-Grissom, H.D. Transcriptomic Insights into the Loss of Vision in Molnar Janos Cave’s Crustaceans. Integr. Comp. Biol. 2018, 58, 452–464. [Google Scholar] [CrossRef]
- Sket, B. Taxonomische Problematik Tier Art Asellus Aquaticus (L.) Rac. Etc. Razpr. Diss. SAZU CI 1965, 4, 1–177. [Google Scholar]
- Nariai, N.; Kojima, K.; Mimori, T.; Kawai, Y.; Nagasaki, M. A Bayesian Approach for Estimating Allele-Specific Expression from RNA-Seq Data with Diploid Genomes. BMC Genom. 2016, 17 (Suppl. 1), 2–5. [Google Scholar] [CrossRef]
- Bastiani, M.J.; de Couet, H.G.; Quinn, J.M.; Karlstrom, R.O.; Kotrla, K.; Goodman, C.S.; Ball, E.E. Position-Specific Expression of the Annulin Protein during Grasshopper Embryogenesis. Dev. Biol. 1992, 154, 129–142. [Google Scholar] [CrossRef]
- Prevorcnik, S.; Blejec, A.S.B. Racial Differentiation in Asellus Aquaticus (L.) (Crustacea: Isopoda: Asellidae. Arch Hydrobiol. 2004, 160, 193–214. [Google Scholar] [CrossRef]
- Jemec, A.; Skufca, D.; Prevorcnik, S.; Fiser, Z.; Zidar, P. Comparative Study of Acetylcholinesterase and Glutathione S-Transferase Activities of Closely Related Cave and Surface Asellus Aquaticus (Isopoda: Crustacea). PLoS ONE 2017, 12, e0176746. [Google Scholar] [CrossRef] [PubMed]
- Fiser, Z.; Prevorcnik, S.; Lozej, N.; Trontelj, P. No Need to Hide in Caves: Shelter-Seeking Behavior of Surface and Cave Ecomorphs of Asellus Aquaticus (Isopoda: Crustacea). Zoology 2019, 134, 58–65. [Google Scholar] [CrossRef]
- Tearle, R.G.; Belote, J.M.; McKeown, M.; Baker, B.S.; Howells, A.J. Cloning and Characterization of the Scarlet Gene of Drosophila Melanogaster. Genetics 1989, 122, 595–606. [Google Scholar]
- Tang, L.; Tong, Y.; Cao, H.; Xie, S.; Yang, Q.; Zhang, F.; Zhu, Q.; Huang, L.; Lu, Q.; Yang, Y.; et al. The MTMR9 rs2293855 Polymorphism is Associated with Glucose Tolerance, Insulin Secretion, Insulin Sensitivity and Increased Risk of Prediabetes. Gene 2014, 546, 150–155. [Google Scholar] [CrossRef]
- Yanagiya, T.; Tanabe, A.; Iida, A.; Saito, S.; Sekine, A.; Takahashi, A.; Tsunoda, T.; Kamohara, S.; Nakata, Y.; Kotani, K.; et al. Association of Single-Nucleotide Polymorphisms in MTMR9 Gene with Obesity. Hum. Mol. Genet. 2007, 16, 3017–3026. [Google Scholar] [CrossRef]
- Mösslacher, F.; Creuzé des Châtelliers, M. Physiological and Behavioural Adaptations of an Epigean and a Hypogean Dwelling Population of Asellus Aquaticus (L.) (Crustacea, Isopoda). Arch. Hydrobiol. 1998, 138, 187–198. [Google Scholar]
- Aspiras, A.C.; Rohner, N.; Martineau, B.; Borowsky, R.L.; Tabin, C.J. Melanocortin 4 Receptor Mutations Contribute to the Adaptation of Cavefish to Nutrient-Poor Conditions. Proc. Natl. Acad. Sci. USA 2015, 112, 9668–9673. [Google Scholar] [CrossRef]
- Riddle, M.R.; Aspiras, A.C.; Gaudenz, K.; Peuss, R.; Sung, J.Y.; Martineau, B.; Peavey, M.; Box, A.C.; Tabin, J.A.; McGaugh, S.; et al. Insulin Resistance in Cavefish as an Adaptation to a Nutrient-Limited Environment. Nature 2018, 555, 647–651. [Google Scholar] [CrossRef]
- Rajput, R.; Ahlawat, P. Prevalence and Predictors of Non-Alcoholic Fatty Liver Disease in Prediabetes. Diabetes Metab. Syndr. 2019, 13, 2957–2960. [Google Scholar] [CrossRef] [PubMed]
- Zidar, P.; Škufca, D.; Prevorčnik, S.; Kalčikova, G.; Jemec Kokalj, A. Energy Reserves in the Water Louse Asellus Aquaticus (Isopoda, Crustacea) from Surface and Cave Populations: Seasonal and Spatial Dynamics. Fundam. Appl. Limonology 2018, 191, 253–265. [Google Scholar] [CrossRef]
- Berning, D.; Adams, H.; Luc, H.; Gross, J.B. In-Frame Indel Mutations in the Genome of the Blind Mexican Cavefish, Astyanax Mexicanus. Genome Biol. Evol. 2019, 11, 2563–2573. [Google Scholar] [CrossRef] [PubMed]
- Gross, J.B.; Borowsky, R.; Tabin, C.J. A Novel Role for Mc1r in the Parallel Evolution of Depigmentation in Independent Populations of the Cavefish Astyanax Mexicanus. PLoS Genet. 2009, 5, e1000326. [Google Scholar] [CrossRef]
- Gross, J.B.; Wilkens, H. Albinism in Phylogenetically and Geographically Distinct Populations of Astyanax Cavefish Arises through the Same Loss-of-Function Oca2 Allele. Heredity 2013, 111, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Protas, M.E.; Hersey, C.; Kochanek, D.; Zhou, Y.; Wilkens, H.; Jeffery, W.R.; Zon, L.I.; Borowsky, R.; Tabin, C.J. Genetic Analysis of Cavefish Reveals Molecular Convergence in the Evolution of Albinism. Nat. Genet. 2006, 38, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Elipot, Y.; Hinaux, H.; Callebert, J.; Launay, J.M.; Blin, M.; Retaux, S. A Mutation in the Enzyme Monoamine Oxidase Explains Part of the Astyanax Cavefish Behavioural Syndrome. Nat. Commun. 2014, 5, 3647. [Google Scholar] [CrossRef]
- Stahl, B.A.; Gross, J.B. Alterations in Mc1r Gene Expression are Associated with Regressive Pigmentation in Astyanax Cavefish. Dev. Genes Evol. 2015, 225, 367–375. [Google Scholar] [CrossRef]
- Wray, G.A. The Evolutionary Significance of Cis-Regulatory Mutations. Nat. Rev. Genet. 2007, 8, 206–216. [Google Scholar] [CrossRef]
- Signor, S.A.; Nuzhdin, S.V. The Evolution of Gene Expression in Cis and Trans. Trends Genet. 2018, 34, 532–544. [Google Scholar] [CrossRef]
- Buchberger, E.; Reis, M.; Lu, T.H.; Posnien, N. Cloudy with a Chance of Insights: Context Dependent Gene Regulation and Implications for Evolutionary Studies. Genes 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H.B. Improving Estimates of Compensatory Cis-Trans Regulatory Divergence. Trends Genet. 2019, 35, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Lawson, H.A.; Cheverud, J.M.; Wolf, J.B. Genomic Imprinting and Parent-of-Origin Effects on Complex Traits. Nat. Rev. Genet. 2013, 14, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Speiser, D.I.; Pankey, M.S.; Zaharoff, A.K.; Battelle, B.A.; Bracken-Grissom, H.D.; Breinholt, J.W.; Bybee, S.M.; Cronin, T.W.; Garm, A.; Lindgren, A.R.; et al. Using Phylogenetically-Informed Annotation (PIA) to Search for Light-Interacting Genes in Transcriptomes from Non-Model Organisms. BMC Bioinform. 2014, 15, 350. [Google Scholar] [CrossRef]
- Carlini, D.B.; Fong, D.W. The Transcriptomes of Cave and Surface Populations of Gammarus Minus (Crustacea: Amphipoda) Provide Evidence for Positive Selection on Cave Downregulated Transcripts. PLoS ONE 2017, 12, e0186173. [Google Scholar] [CrossRef]
- Huang, Z.; Titus, T.; Postlethwait, J.H.; Meng, F. Eye Degeneration and Loss of Otx5b Expression in the Cavefish Sinocyclocheilus Tileihornes. J. Mol. Evol. 2019, 87, 199–208. [Google Scholar] [CrossRef]
- Friedrich, M.; Chen, R.; Daines, B.; Bao, R.; Caravas, J.; Rai, P.K.; Zagmajster, M.; Peck, S.B. Phototransduction and Clock Gene Expression in the Troglobiont Beetle Ptomaphagus Hirtus of Mammoth Cave. J. Exp. Biol. 2011, 214, 3532–3541. [Google Scholar] [CrossRef]
- Tierney, S.M.; Langille, B.; Humphreys, W.F.; Austin, A.D.; Cooper, S.J.B. Massive Parallel Regression: A Precis of Genetic Mechanisms for Vision Loss in Diving Beetles. Integr. Comp. Biol. 2018, 58, 465–479. [Google Scholar] [CrossRef]
- Lefebure, T.; Morvan, C.; Malard, F.; Francois, C.; Konecny-Dupre, L.; Gueguen, L.; Weiss-Gayet, M.; Seguin-Orlando, A.; Ermini, L.; Sarkissian, C.; et al. Less Effective Selection Leads to Larger Genomes. Genome Res. 2017, 27, 1016–1028. [Google Scholar] [CrossRef]
- Konec, M.; Prevorcnik, S.; Sarbu, S.M.; Verovnik, R.; Trontelj, P. Parallels between Two Geographically and Ecologically Disparate Cave Invasions by the Same Species, Asellus Aquaticus (Isopoda, Crustacea). J. Evol. Biol. 2015, 28, 864–875. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Surface Morphs | Cave Morphs | Hybrids | |
|---|---|---|---|
| Sequence Read Summary | |||
| Total Assembled Reads | 155039720 | 164487662 | 132336702 |
| Total Unassembled Reads | 83386227 | 109047509 | 84947885 |
| Total Reads Excluded by Sampling | 126373422 | 87592389 | 176811792 |
| Total Number of Reads | 364799369 | 361127560 | 394096379 |
| Transcript Summary | |||
| Total number of Transcripts | 113432 | 119569 | 143962 |
| Average Length of Assembled Transcripts | 1061 | 1069 | 952 |
| Assembled Transcripts >1kb | 49,233 | 51,822 | 52,390 |
| Assembly Time | 50.7 h | 54.7 h | 54.2 h |
| Surface Morphs | Cave Morphs | Hybrids | |
| Tribolium Genome Database | |||
| Total Number of Transcripts | 113,432 | 119,569 | 143,962 |
| Assembled Transcripts >1kb | 49,233 | 51,822 | 52,390 |
| No BLAST hits | 28,648 | 30,340 | 30,709 |
| Ribosomal sequences | 518 | 749 | 712 |
| Mitochondrial sequences | 880 | 734 | 973 |
| Total number of annotated sequences | 19,187 | 19,999 | 19,996 |
| Surface Morphs | Cave Morphs | Hybrids | |
| SwissProt Database | |||
| Total Number of Transcripts | 113,432 | 119,569 | 143,962 |
| Assembled Transcripts >1kb | 49,233 | 51,822 | 52,390 |
| No BLAST hits | 29,918 | 31,928 | 32,157 |
| Ribosomal sequences | 603 | 624 | 839 |
| Mitochondrial sequences | 986 | 992 | 1066 |
| Total number of annotated sequences | 17,726 | 18,278 | 18,328 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gross, J.B.; Sun, D.A.; Carlson, B.M.; Brodo-Abo, S.; Protas, M.E. Developmental Transcriptomic Analysis of the Cave-Dwelling Crustacean, Asellus aquaticus. Genes 2020, 11, 42. https://doi.org/10.3390/genes11010042
Gross JB, Sun DA, Carlson BM, Brodo-Abo S, Protas ME. Developmental Transcriptomic Analysis of the Cave-Dwelling Crustacean, Asellus aquaticus. Genes. 2020; 11(1):42. https://doi.org/10.3390/genes11010042
Chicago/Turabian StyleGross, Joshua B., Dennis A. Sun, Brian M. Carlson, Sivan Brodo-Abo, and Meredith E. Protas. 2020. "Developmental Transcriptomic Analysis of the Cave-Dwelling Crustacean, Asellus aquaticus" Genes 11, no. 1: 42. https://doi.org/10.3390/genes11010042
APA StyleGross, J. B., Sun, D. A., Carlson, B. M., Brodo-Abo, S., & Protas, M. E. (2020). Developmental Transcriptomic Analysis of the Cave-Dwelling Crustacean, Asellus aquaticus. Genes, 11(1), 42. https://doi.org/10.3390/genes11010042