Local Epigenomic Data are more Informative than Local Genome Sequence Data in Predicting Enhancer-Promoter Interactions Using Neural Networks


Abstract
1. Introduction
2. Materials and Methods
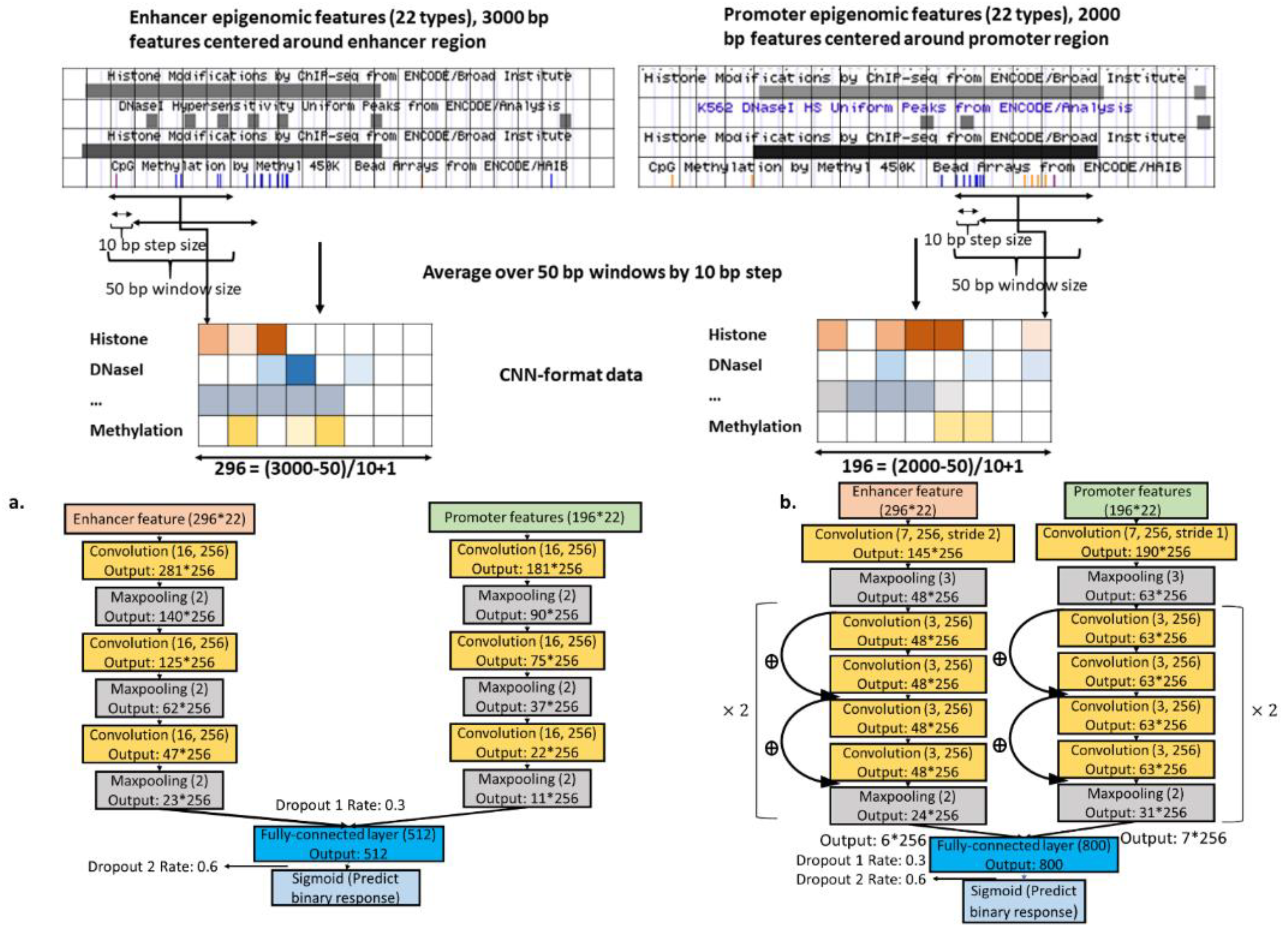
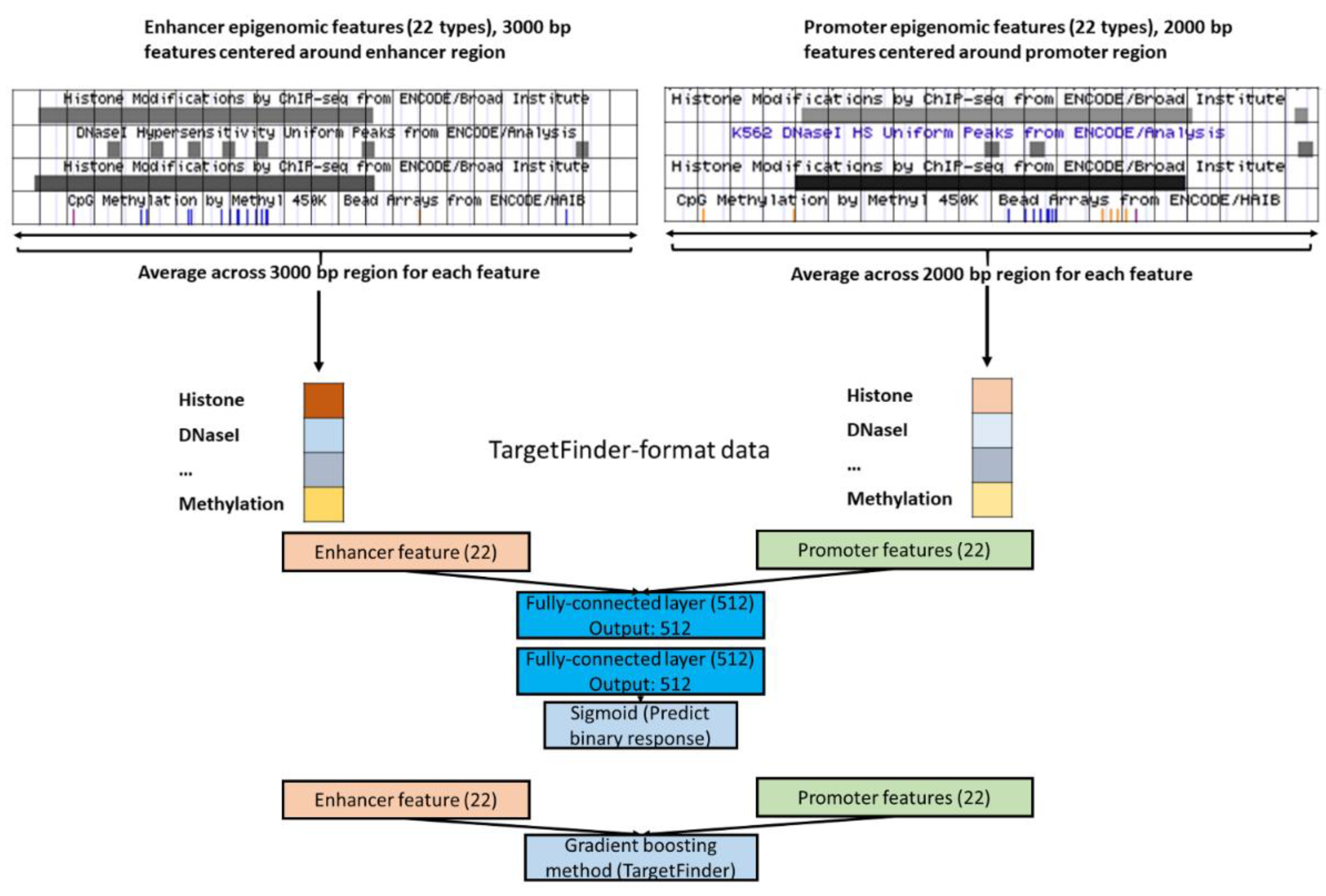
2.1. Data
2.2. Designing Neural Networks for Utilizing Enhancer and Promoter Features
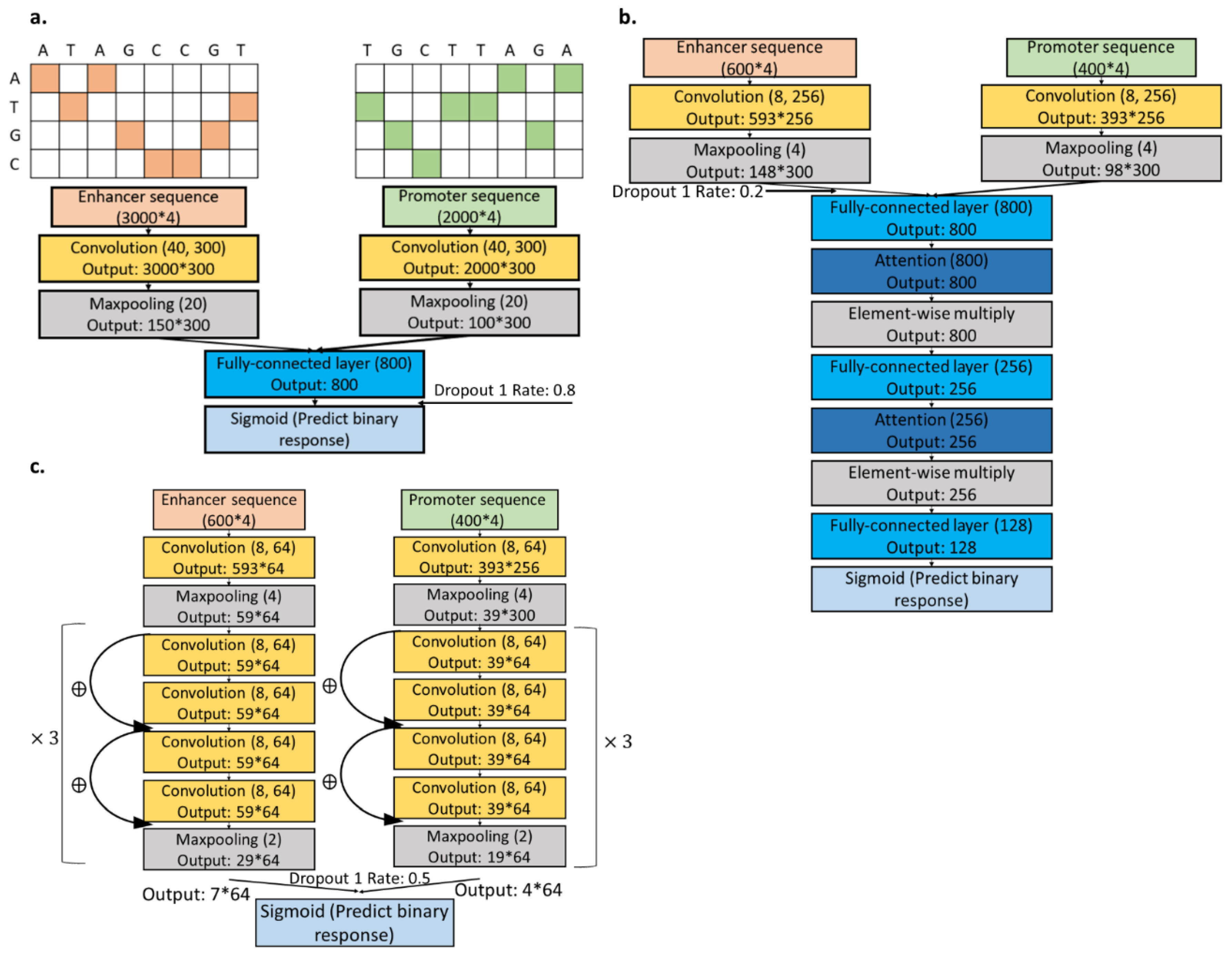
2.3. Sequence CNNs.
2.4. Epigenomics CNNs
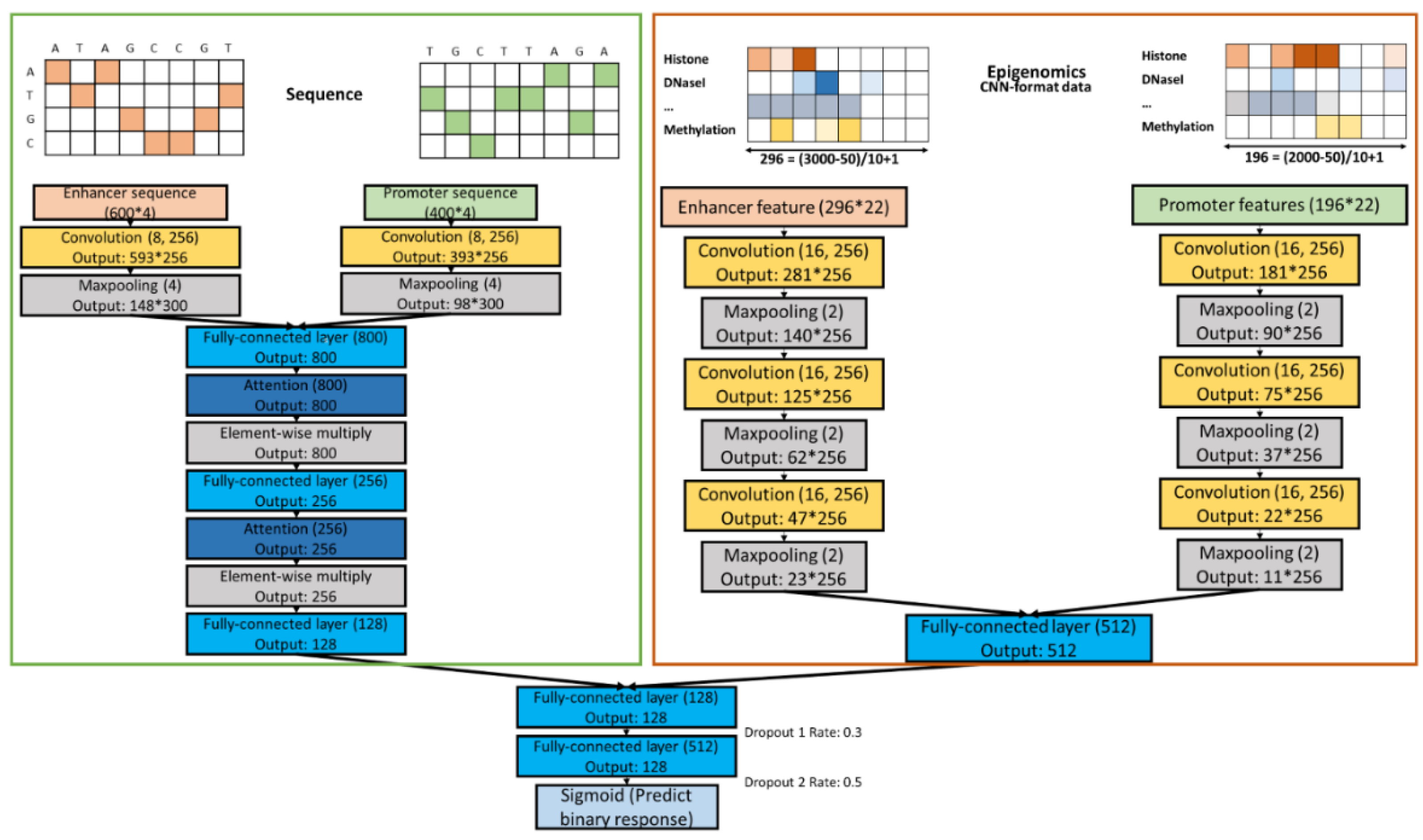
2.5. Combined Models
2.6. Designing FNNs.
2.7. CNN Training in an Imbalanced-Class Scenario
2.8. Evaluating Model Performance
2.9. Implementation and Code Availability
3. Results
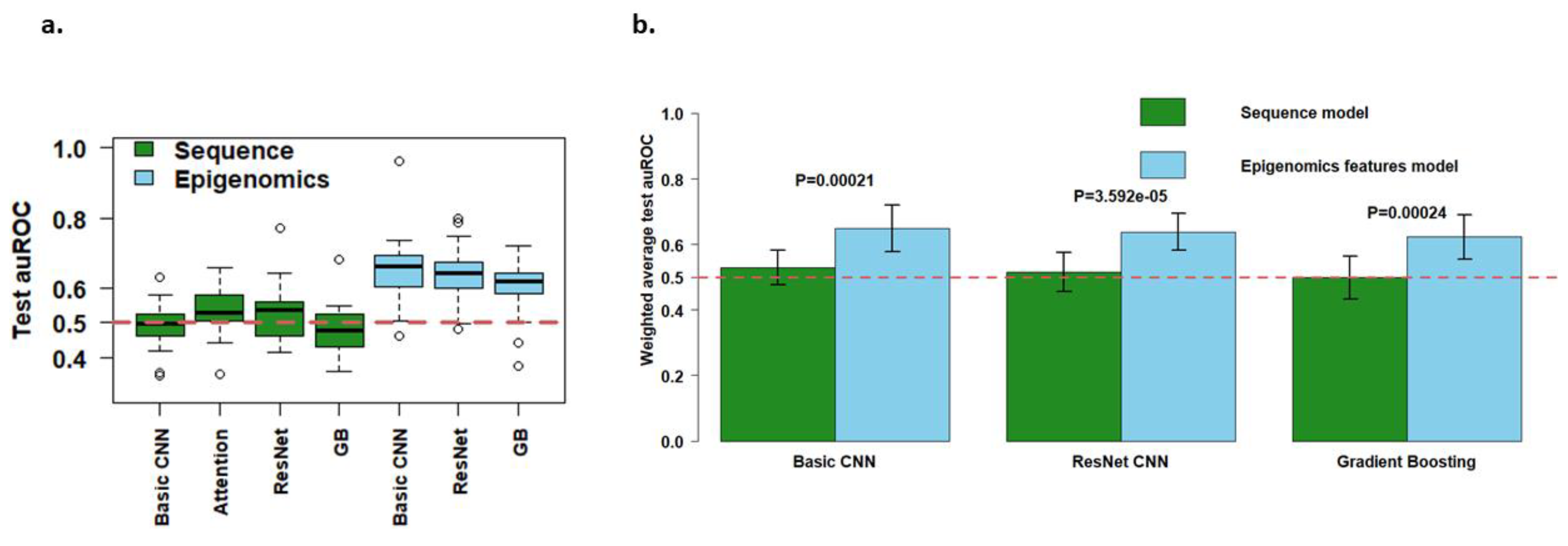
3.1. Local Epigenomic Features Were more Informative than Local Sequence Data in Predicting EPIs
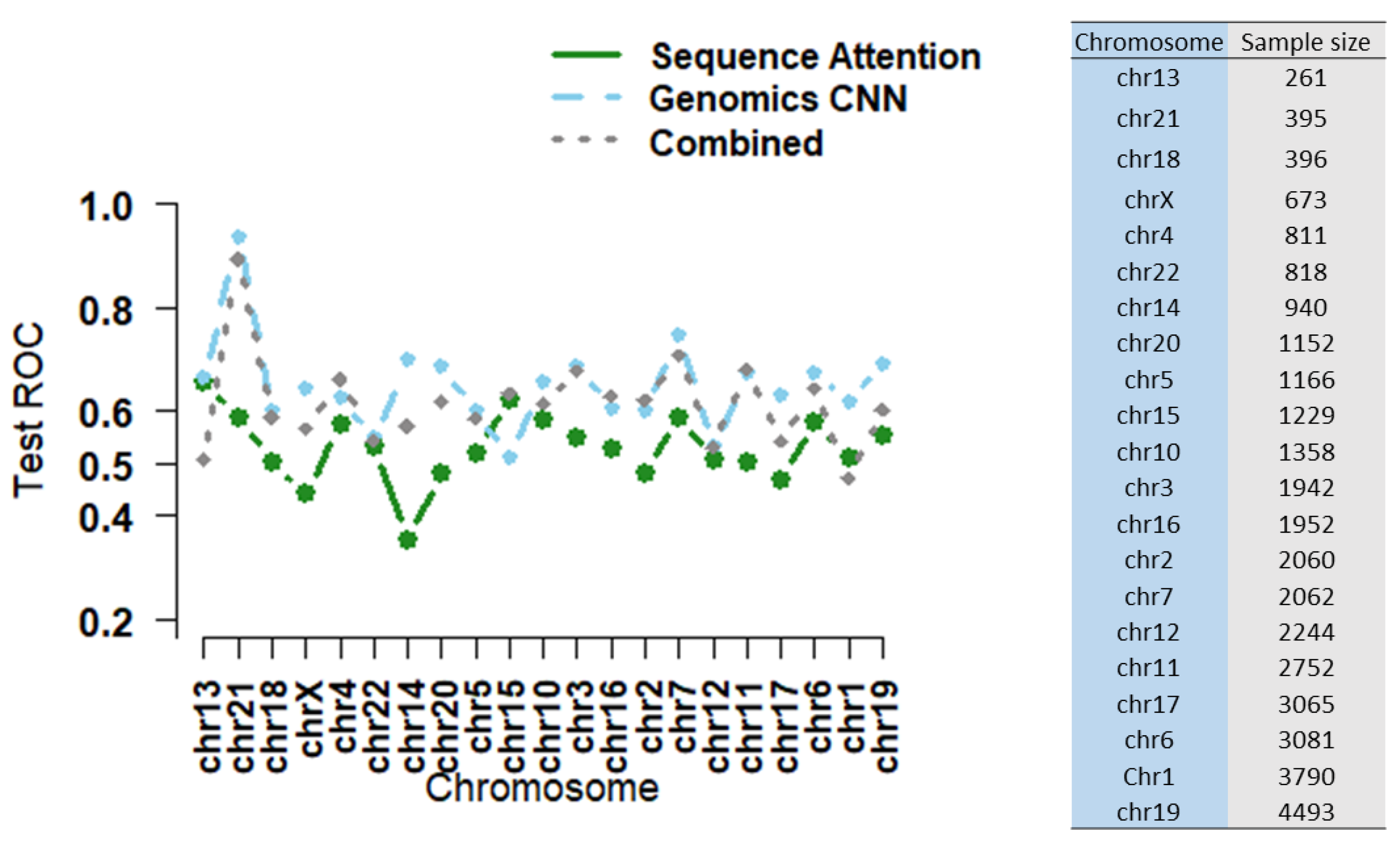
3.2. Combining Epigenomic and Sequence Data: Do We Gain Additional Information?
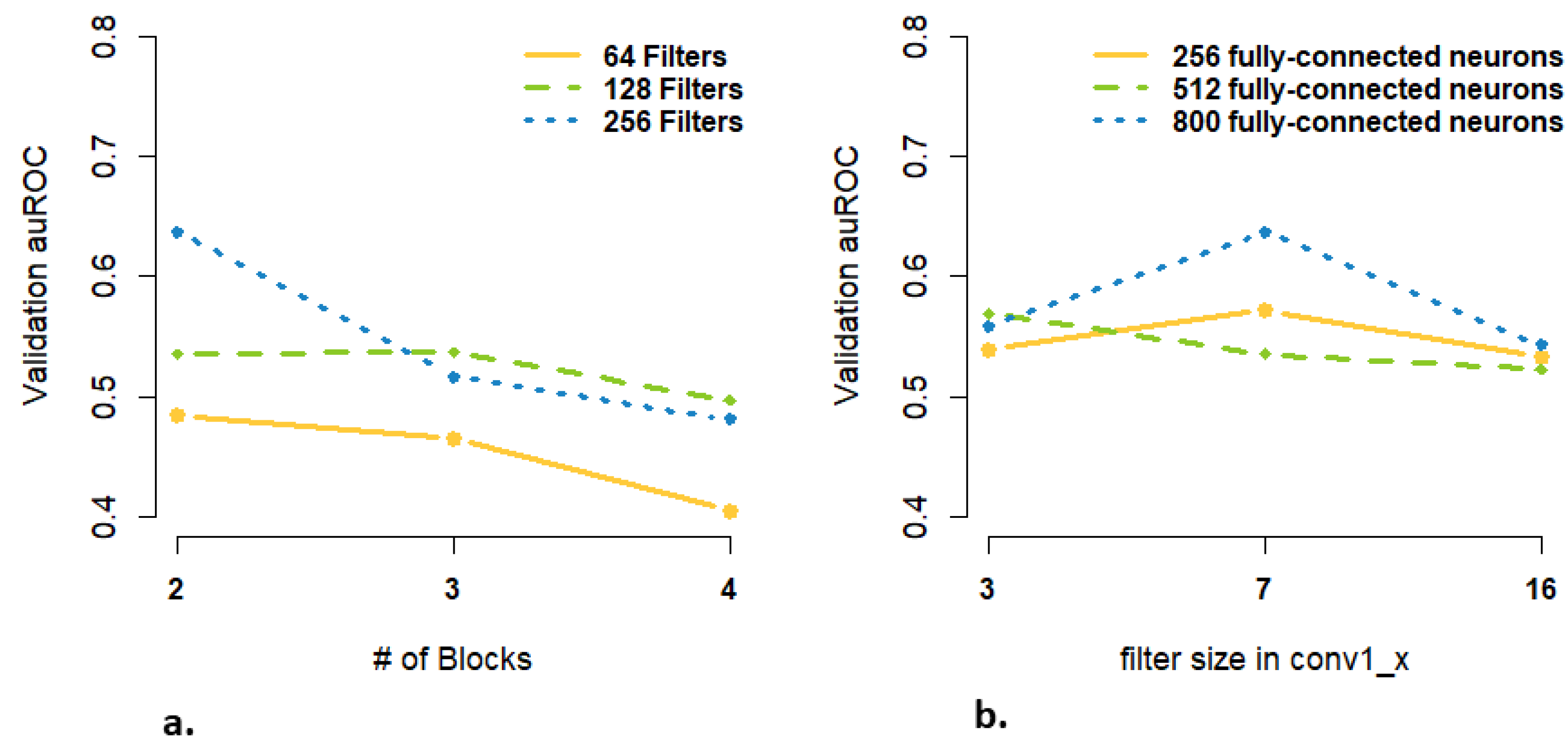
3.3. Are more Complex Structures and more Parameters Needed for High-Dimensional Data Input?
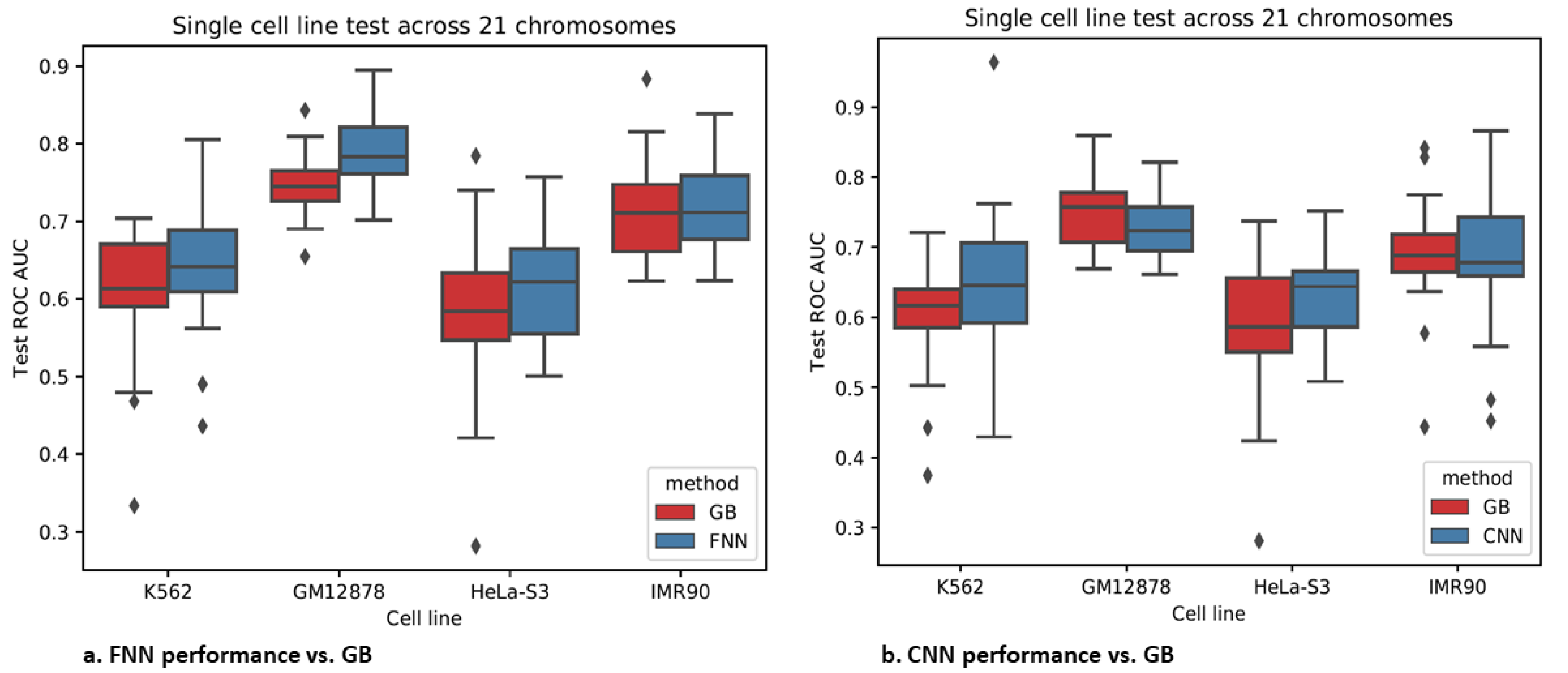
3.4. Epigenomics Feed-Forward Neural Networks (FNNs) Performed Better than Gradient Boosting
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schoenfelder, S.; Fraser, P. Long-range enhancer-promoter contacts in gene expression control. Nat. Rev. Genet. 2019, 20, 437–455. [Google Scholar] [CrossRef] [PubMed]
- Won, H.; de La Torre-Ubieta, L.; Stein, J.L.; Parikshak, N.N.; Huang, J.; Opland, C.K.; Gandal, M.J.; Sutton, G.J.; Hormozdiari, F.; Lu, D.; et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 2016, 538, 523. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Pan, W. Integration of Enhancer-Promoter Interactions with GWAS Summary Results Identifies Novel Schizophrenia-Associated Genes and Pathways. Genetics 2018, 209, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Dekker, J.; Rippe, K.; Dekker, M.; Kleckner, N. Capturing chromosome conformation. Science 2002, 295, 1306–1311. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Ruan, X.; Auerbach, R.K.; Sandhu, K.S.; Zheng, M.; Wang, P.; Poh, H.M.; Goh, Y.; Lim, J.; Zhang, J.; et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 2012, 148, 84–98. [Google Scholar] [CrossRef]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Sanyal, A.; Lajoie, B.R.; Jain, G.; Dekker, J. The long-range interaction landscape of gene promoters. Nature 2012, 489, 109. [Google Scholar] [CrossRef]
- Cao, Q.; Anyansi, C.; Hu, X.; Xu, L.; Xiong, L.; Tang, W.; Mok, M.T.; Cheng, C.; Fan, X.; Gerstein, M.; et al. Reconstruction of enhancer-target networks in 935 samples of human primary cells, tissues and cell lines. Nat. Genet. 2017, 49, 1428. [Google Scholar] [CrossRef]
- He, B.; Chen, C.; Teng, L.; Tan, K. Global view of enhancer-promoter interactome in human cells. Proc. Natl. Acad. Sci. USA 2014, 111, E2191–E2199. [Google Scholar] [CrossRef]
- Roy, S.; Siahpirani, A.F.; Chasman, D.; Knaack, S.; Ay, F.; Stewart, R.; Wilson, M.; Sridharan, R. A predictive modeling approach for cell line-specific long-range regulatory interactions. Nucleic Acids Res. 2015, 43, 8694–8712. [Google Scholar] [CrossRef]
- Whalen, S.; Truty, R.M.; Pollard, K.S. Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat. Genet. 2016, 48, 488. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, R.; Singh, S.; Ma, J. Exploiting sequence-based features for predicting enhancer-promoter interactions. Bioinformatics 2017, 33, I252–I260. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Yang, Y.; Poczos, B.; Ma, J. Predicting enhancer-promoter interaction from genomic sequence with deep neural networks. bioRxiv 2016, 085241. [Google Scholar] [CrossRef]
- Zhuang, Z.; Shen, X.T.; Pan, W. A simple convolutional neural network for prediction of enhancer-promoter interactions with DNA sequence data. Bioinformatics 2019, 35, 2899–2906. [Google Scholar] [CrossRef]
- Geirhos, R.; Rubisch, P.; Michaelis, C.; Bethge, M.; Wichmann, F.A.; Brendel, W. ImageNet-trained CNNs are biased towards texture; increasing shape bias improves accuracy and robustness. arXiv 2018, arXiv:1811.12231. [Google Scholar]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. Imagenet classification with deep convolutional neural networks. In Advances in Neural Information Processing Systems; Pereira, F., Burges, C.J.C., Bottou, L., Weinberger, K.Q., Eds.; Curran Associates, Inc.: Lake Tahoe, NV, USA, 2012; pp. 1097–1105. [Google Scholar]
- Luo, X.; Chi, W.; Deng, M. Deepprune: Learning efficient and interpretable convolutional networks through weight pruning for predicting DNA-protein binding. bioRxiv 2019, 729566. [Google Scholar] [CrossRef]
- Cao, F.; Fullwood, M.J. Inflated performance measures in enhancer-promoter interaction-prediction methods. Nat. Genet. 2019, 51, 1196–1198. [Google Scholar] [CrossRef]
- Xi, W.; Beer, M.A. Local epigenomic state cannot discriminate interacting and non-interacting enhancer-promoter pairs with high accuracy. PLoS Comput. Biol. 2018, 14, e1006625. [Google Scholar] [CrossRef]
- Li, W.R.; Wong, W.H.; Jiang, R. DeepTACT: Predicting 3D chromatin contacts via bootstrapping deep learning. Nucleic Acids Res. 2019, 47, e60. [Google Scholar] [CrossRef]
- Kong, Y.; Yu, T. A graph-embedded deep feedforward network for disease outcome classification and feature selection using gene expression data. Bioinformatics 2018, 34, 3727–3737. [Google Scholar] [CrossRef]
- Liang, F.; Li, Q.; Zhou, L. Bayesian neural networks for selection of drug sensitive genes. J. Am. Stat. Assoc. 2018, 113, 955–972. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Stamatoyannopoulos, J.A.; Costello, J.F.; Ren, B.; Milosavljevic, A.; Meissner, A.; Kellis, M.; Marra, M.A.; Beaudet, A.L.; Ecker, J.R.; et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat. Biotechnol. 2010, 28, 1045–1048. [Google Scholar] [CrossRef] [PubMed]
- Encode Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Ira, M.H. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Bahdanau, D.; Cho, K.; Bengio, Y. Neural machine translation by jointly learning to align and translate. arXiv 2014, arXiv:1409.0473. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep residual learning for image recognition. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Identity mappings in deep residual networks. In Proceedings of the European Conference on Computer Vision, Amsterdam, The Netherlands, 8–16 October 2016; pp. 630–645. [Google Scholar]
- Sabour, S.; Frosst, N.; Hinton, G.E. Dynamic routing between capsules. In Advances in Neural Information Processing Systems; Guyon, I., Luxburg, U.V., Bengio, S., Wallach, H., Fergus, R., Vishwanathan, S., Garnett, R., Eds.; Curran Associates, Inc.: Long Beach, CA, USA, 2017; pp. 3856–3866. [Google Scholar]
- Szegedy, C.; Ioffe, S.; Vanhoucke, V.; Alemi, A.A. Inception-v4, inception-resnet and the impact of residual connections on learning. In Proceedings of the Thirty-First AAAI Conference on Artificial Intelligence, San Francisco, CA, USA, 4–9 February 2017. [Google Scholar]
- Ioffe, S.; Szegedy, C. Batch normalization: Accelerating deep network training by reducing internal covariate shift. arXiv 2015, arXiv:1502.03167. [Google Scholar]
- Glorot, X.; Bengio, Y. Understanding the difficulty of training deep feedforward neural networks. In Proceedings of the Thirteenth International Conference on Artificial Intelligence and Statistics, Chia Laguna, Italy, 13–15 May 2010; pp. 249–256. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Delving deep into rectifiers: Surpassing human-level performance on imagenet classification. In Proceedings of the IEEE International Conference on Computer Vision, Santiago, Chile, 7–13 December 2015; pp. 1026–1034. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A Simple Way to Prevent Neural Networks from Overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Jing, F.; Zhang, S.; Cao, Z.; Zhang, S. An integrative framework for combining sequence and epigenomic data to predict transcription factor binding sites using deep learning. IEEE ACM Trans. Comput. Biol. Bioinform. 2019. [Google Scholar] [CrossRef]
- Nair, S.; Kim, D.S.; Perricone, J.; Kundaje, A. Integrating regulatory DNA sequence and gene expression to predict genome-wide chromatin accessibility across cellular contexts. Bioinformatics 2019, 35, i108–i116. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Schwessinger, R.; Gosden, M.; Downes, D.; Brown, R.; Telenius, J.; Teh, Y.W.; Lunter, G.; Hughes, J.R. DeepC: Predicting chromatin interactions using megabase scaled deep neural networks and transfer learning. bioRxiv 2019, 724005. [Google Scholar] [CrossRef]
- Whalen, S.; Pollard, K.S. Reply to ‘Inflated performance measures in enhancer-promoter interaction-prediction methods’. Nat. Genet. 2019, 51, 1198–1200. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Batch Size | Learning Rate | L2 Weight Decay in the Convolution | Dropout 1 | Dropout 2 | |
|---|---|---|---|---|---|
| Sequence | |||||
| Zhuang et al., 2019 [14] (Basic CNN) | 64 | 1.0 × 10−5 | 1.0 × 10−6 | 0.2 | 0.8 |
| Attention CNN | 64 | 1.0 × 10−5 | 2.0 × 10−5 | 0.2 | None |
| ResNet CNN | 200 | 1.0 × 10−5 | 2.0 × 10−5 | 0.5 | None |
| Epigenomics | |||||
| Basic CNN | 200 | 1.0 × 10−6 | 1.0 × 10−5 | 0.3 | 0.6 |
| ResNet CNN | 64 | 1.0 × 10−3 | 1.0 × 10−5 | 0.3 | 0.6 |
| FNN | 200 | 1.0 × 10−6 | 1.0 × 10−5 | 0.7 | None |
| Combined model | 200 | 1.0 × 10−5 | 1.0 × 10−6 | 0.3 | 0.5 |
| Block Name | Simple/Basic CNN | Attention | ResNet without Fully-Connected Layer |
|---|---|---|---|
| Input | |||
| conv1_x | |||
| conv2_x | |||
| conv3_x | |||
| conv4_x | |||
| Concatenate branches | 800-d fc, sigmoid | ||
| # of parameters | 60,100,402 | 51,345,538 | 605,452 |
| Block Name | Basic CNN (2 Branches) | Basic CNN (1 Branch) | ResNet with Fully-Connected (fc) Layer | ResNet without Fully-Connected (fc) Layer |
|---|---|---|---|---|
| Input | ||||
| conv1_x | ||||
| conv2_x | ||||
| conv3_x | ||||
| conv4_x | ||||
| conv5_x | ||||
| Concatenate branches | ||||
| # of parameters | 8,838,145 | 12,244,481 | 5,915,841 | 1,625,985 |
| Epigenomics Model | Mean AUROC | Standard Deviation of AUROC | Sequence Model | Mean AUROC | Standard Deviation of AUROC |
|---|---|---|---|---|---|
| Simple convolution/Basic sequence CNN (Zhuang et al., 2019 [14]; original region) | 0.500 | 0.0500 | Basic CNN | 0.648 | 0.0704 |
| Attention CNN (central region) | 0.529 | 0.0533 | ResNet CNN | 0.638 | 0.0568 |
| ResNet CNN (central region) | 0.515 | 0.0596 | Gradient Boosting | 0.622 | 0.0676 |
| Gradient Boosting (central region) | 0.494 | 0.0557 | Combined model | 0.603 | 0.0703 |
| Gradient Boosting (original region) | 0.499 | 0.0654 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, M.; Zhuang, Z.; Pan, W. Local Epigenomic Data are more Informative than Local Genome Sequence Data in Predicting Enhancer-Promoter Interactions Using Neural Networks. Genes 2020, 11, 41. https://doi.org/10.3390/genes11010041
Xiao M, Zhuang Z, Pan W. Local Epigenomic Data are more Informative than Local Genome Sequence Data in Predicting Enhancer-Promoter Interactions Using Neural Networks. Genes. 2020; 11(1):41. https://doi.org/10.3390/genes11010041
Chicago/Turabian StyleXiao, Mengli, Zhong Zhuang, and Wei Pan. 2020. "Local Epigenomic Data are more Informative than Local Genome Sequence Data in Predicting Enhancer-Promoter Interactions Using Neural Networks" Genes 11, no. 1: 41. https://doi.org/10.3390/genes11010041
APA StyleXiao, M., Zhuang, Z., & Pan, W. (2020). Local Epigenomic Data are more Informative than Local Genome Sequence Data in Predicting Enhancer-Promoter Interactions Using Neural Networks. Genes, 11(1), 41. https://doi.org/10.3390/genes11010041